Uncategorized Friday, 2017/12/01
(Continued)
5. On average, one to ten mutations are sufficient to promote cancer
Iñigo Martincorena, Keiran M. Raine, Moritz Gerstung et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell, Published online: October 19, 2017, doi:10.1016/j.cell.2017.09.042
[caption id="attachment_738" align="aligncenter" width="565"]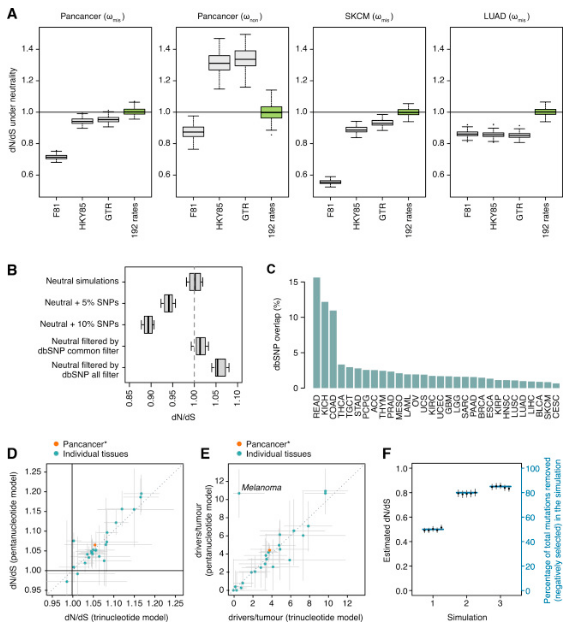 Universal Patterns of Selection in Cancer and Somatic Tissues[/caption]
In a study of more than 7,500 tumors of 29 cancer types, researchers from the Wellcome Trust Sanger Institute, the European Bioinformatics Institute, and the Francis-Crick Institute unbiased evaluated the number of mutations needed to produce cancer for the first time. They have demonstrated that on average, 1 to 10 driver mutations are necessary for cancer production by improving one technique in the field of evolution. These results also confirm that there is a considerable difference in the number of mutations that contribute to the development of cancer between different cancers.
Universal Patterns of Selection in Cancer and Somatic Tissues[/caption]
In a study of more than 7,500 tumors of 29 cancer types, researchers from the Wellcome Trust Sanger Institute, the European Bioinformatics Institute, and the Francis-Crick Institute unbiased evaluated the number of mutations needed to produce cancer for the first time. They have demonstrated that on average, 1 to 10 driver mutations are necessary for cancer production by improving one technique in the field of evolution. These results also confirm that there is a considerable difference in the number of mutations that contribute to the development of cancer between different cancers.
In the study, these researchers quantified the natural selection of 7664 tumors of 29 different cancer types from an evolutionary perspective.
A prominent finding of this study is that cells in the body have great tolerance for mutations. This is surprising because mutations that individuals inherit from their parents are often less well tolerated and usually disappear from humans over time. However, in the body's cells, almost all mutations persist as cancer develops, but it does not affect cell survival.
The researchers also register the major oncogenes that cause 29 different cancer types. They found several new oncogenes and determined the completeness of the current oncogene list.
Dr. Campbell said, "We solved the longstanding question that has been debated since the 1950s: how many mutations a normal cell needs to transform into a cancer cell? The answer is a small fraction, for example, on average approximately four mutations in each patient triggers liver cancer, whereas colorectal cancer usually requires about 10 driver mutations. "
6. Reveal the molecular mechanism of cytoplasmic DNA-induced inflammatory responses in human cells
Moritz M. Gaidt, Thomas S. Ebert, Dhruv Chauhan, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell (2017). DOI: 10.1016/j.cell.2017.09.039
[caption id="attachment_739" align="aligncenter" width="377"]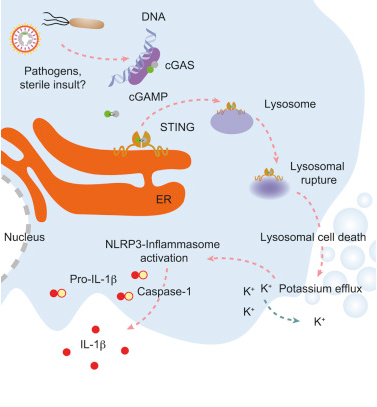 The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3 [/caption]
Recently, a study published in the international magazine Cell reported that researchers from the University of Munich have elucidated the molecular mechanism of cytoplasmic DNA-induced inflammation in human cells. It is noteworthy that the signal network involved may be different with the mouse body.
The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3 [/caption]
Recently, a study published in the international magazine Cell reported that researchers from the University of Munich have elucidated the molecular mechanism of cytoplasmic DNA-induced inflammation in human cells. It is noteworthy that the signal network involved may be different with the mouse body.
In eukaryotes, DNA is confined in the nucleus, and the presence of DNA in the cytoplasm is a signal that cells are in a very dangerous situation. Cytoplasmic DNA may originate from viruses or bacteria, which can suggest an infection or damage from endogenous source or tissue. Therefore, recognition of the cytoplasmic DNA by the innate immune system induces a wide range of inflammatory confrontations and body defense. In this study, researchers have elucidated a specific mechanism by which can promote innate immune systems in human cells to recognize such specific DNA and induce inflammatory responses.
Hornung, the researcher, explains that their recent study of human myeloid cells found that the receptor plays a key role in the process described above, which identifies foreign or ectopic DNA in a different way. Compared with mice, the inflammasome molecules of human myeloid cells can be activated by a recognition mechanism called cGAS-STING that can help induce an innate immune response to the appearance of viral DNA.
Now researchers have found that activation of the cGAS-STING pathway can induce programmed cell death, which does not depend on the antiviral response. STING proteins induce lysosomal fragmentation when the cGAS-STING pathway is activated beyond a certain threshold. Then the cell damage activates the inflammasome, which secretes interleukin-1 as an emergency distress signal, via which the dead cell sends an alert to nearby cells, recruiting immune cells to the emergency site.
7. Reveal the Mechanism of 3D Packaging of DNA Regulates Cell Identity
Andrey Poleshko, Parisha P. Shah, Mudit Gupta et al. Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell, Published online: October 12, 2017, doi:10.1016/j.cell.2017.09.018
[caption id="attachment_740" align="aligncenter" width="374"]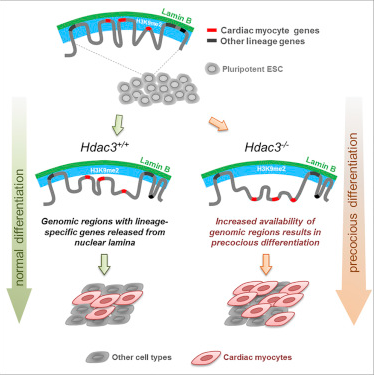 Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction[/caption]
The basic mechanism by which a cell retains its identity (such as becoming a muscle cell or nerve cell) is not fully understood. Many diseases, such as cancer, are associated with the wrong path of development in which cells are selected during maturation. In a new study, researchers from Perelman College of Medicine, University of Pennsylvania, and Icahn County Mount Sinai, proposed that the ability of stem cells to differentiate into cardiomyocytes (and other cell types) depends on which regions of the genome are being activated, which is controlled by the location of DNA in the nucleus. Relevant findings were published online in the Cell journal on October 12, 2017. The paper was written by Jonathan A. Epstein, Ph.D., executive vice president and chief scientist at Perelman Medical School, University of Pennsylvania, and Rajan Jain, assistant professor of cardiovascular medicine at Perelman Medical School, University of Pennsylvania.
Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction[/caption]
The basic mechanism by which a cell retains its identity (such as becoming a muscle cell or nerve cell) is not fully understood. Many diseases, such as cancer, are associated with the wrong path of development in which cells are selected during maturation. In a new study, researchers from Perelman College of Medicine, University of Pennsylvania, and Icahn County Mount Sinai, proposed that the ability of stem cells to differentiate into cardiomyocytes (and other cell types) depends on which regions of the genome are being activated, which is controlled by the location of DNA in the nucleus. Relevant findings were published online in the Cell journal on October 12, 2017. The paper was written by Jonathan A. Epstein, Ph.D., executive vice president and chief scientist at Perelman Medical School, University of Pennsylvania, and Rajan Jain, assistant professor of cardiovascular medicine at Perelman Medical School, University of Pennsylvania.
The study also suggests that understanding the ways in which stem cells rapidly differentiate during maturation can have a major impact on regenerative medicine. Some regions of the genome are not expressed because they are tightly packed on the inner membrane of the nucleus. These isolated and silent DNA regions are called laminated domains (LADs). The study suggests that these specific DNA-silencing regions at the nucleus edge help identify cells’ type. For example, if LAD silences a neuronal gene, the cell does not become a neuron. On the contrary, if the heart cell genes are released to be expressed, as happens during heart development, then these cells become cardiomyocytes. Cell biologists have known for years that some DNA is found near the inner nuclear membrane, but the function of this localization is unclear. Jain says, "Our research suggests that cells identify their identity by storing key genes and necessary programs which could transform it into another cell types in hard-to-reach areas. In other words, cells retain their identity by suppressing itself transform into another identity. "
The researchers found that an epigenetic enzyme called histone deacetylase (HDAC3) attaches DNA to the edges of the nucleus. Jain said, "We asked: will careful accessible control of DNA lead to cells becoming desirable cell type?" When they removed HDAC3 from stem cells during cardiocyte differentiation, they released genes that contain heart-specific genes DNA region, allowing these genes to be activated, leading to premature and over-rapid differentiation.
8. Genome sequencing reveals new genetic variations in autism
Tychele N. Turner, Bradley P. Coe, Diane E. Dickel et al. Genomic Patterns of De Novo Mutation in Simplex Autism. Cell, Published online: September 28, 2017, doi:10.1016/j.cell.2017.08.047
[caption id="attachment_741" align="aligncenter" width="386"]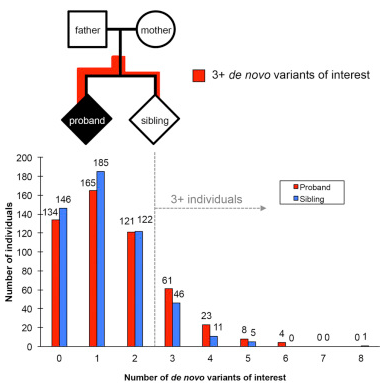 Genomic Patterns of De Novo Mutation in Simplex Autism[/caption]
Today, the genomic pattern found in children with autism - a complete set of genetic instructions in the cell - reveals a new genetic trait of the disease. This feature helps to explain the absence of other genetic markers of autism cases. Relevant findings were published online in the Cell journal on September 28, 2017, and the article entitled "Genomic Patterns of De Novo Mutation in Simplex Autism".
Genomic Patterns of De Novo Mutation in Simplex Autism[/caption]
Today, the genomic pattern found in children with autism - a complete set of genetic instructions in the cell - reveals a new genetic trait of the disease. This feature helps to explain the absence of other genetic markers of autism cases. Relevant findings were published online in the Cell journal on September 28, 2017, and the article entitled "Genomic Patterns of De Novo Mutation in Simplex Autism".
The researchers sequenced 516 children’s genome with autism who did not have a family history of autism (genetic abnormalities were not detected in these children using current testing methods). They also conducted genome sequencing of the parents of these children and one sibling (a total of 2,064 people) who were not affected by the disease. This genetic information is stored in the Simons Simplex Collection (SSC) database.
The Eichler team identified genetic changes that led to a disruption of gene function and changes in protein expression, as well as gene deletions. These missing fragments are too small to be detected using current testing methods. They also found changes in genomic regions which did not contain genes but could lead to gene activation. Turner said: “Eichler and colleagues have tagged all these changes in the SSC database so that others can use the findings as a resource”.
The researchers then compared the number of genomic variations in autistic children and their siblings who were not affected by the disease. They found that children with autism are significantly more likely to have three or more different types of genetic variation. Eichler said: “This suggests that sporadic combinations of genetic variations may lead to autism. However,” he stressed that. “Scientists need to replicate these findings in more families before using specific genes or combinations of genes for diagnosis”.
(to be continued...)