Cerebellar ataxia can occur as a result of many diseases and presents with symptoms of an inability to coordinate balance, gait, extremity and eye movements. There are many causes of cerebellar ataxia including, among others, autoimmunity to Purkinje cells or other neural cells in the cerebellum, CNS vacuities, multiple sclerosis, infection, bleeding, infarction, tumors, direct injury, toxins, genetic disorders, and an association with statin use.
Inherited (genetic) forms of ataxia must be distinguished from the many acquired (non-genetic) causes of ataxia. The genetic forms of ataxia are diagnosed by family history, physical examination, neuroimaging, and molecular genetic testing. The hereditary ataxias are categorized by mode of inheritance and gene (or chromosome locus) in which pathogenic variants occur.
Recently, a new letter from Nature found that “XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia”.
 Fig1. XRCC1 mutations are associated with cerebellar ataxia, ocular motor apraxia, and axonal neuropathy
Fig1. XRCC1 mutations are associated with cerebellar ataxia, ocular motor apraxia, and axonal neuropathy
A woman was diagnosed at age 41 with cerebellar atrophy, gait and limb ataxia, ocular motor apraxia, and peripheral neuropathy, which was noticed at 28 years. After ruling out more than ten known spinocerebellar ataxias by genetic and metabolic screening, exome sequencing of the proband identified compound heterozygous mutations in XRCC1.
The pathogenic impact of XRCC1 mutations was detected by immunofluorescence and WB. The level of XRCC1 was significant reduced in patient primary fibroblasts compared with wild-type (WT), and not was not measurably higher than in human RPE-1 cells in which XRCC1 was deleted by clustered, regularly interspaced short palindromic repeats (CRISPR)-Cas9. Levels of DNA ligase IIIα (Lig3α) were also greatly reduced (by >80%) in patient cells, consistent with the established impact of XRCC1 on the cellular stability of this partner protein.
 Fig2. Mutations in patient-derived XRCC1 reduce XRCC1 levels
Fig2. Mutations in patient-derived XRCC1 reduce XRCC1 levels
After treatment with H2O2, a physiological source of oxidative single-strand breaks (SSBs), little or no XRCC1 recruitment into chromatin was detected in XRCC1-patient fibroblasts compared with WT, in which XRCC1 was rapidly recruited into global nuclear chromatin. Similar results were observed after treatment with camptothecin (CPT), a topoisomerase poison that induces SSBs triggered by abortive topoisomerase I activity.
XRCC1 is a molecular scaffold protein that assembles multiprotein complexes involved in DNA single-strand break repair. SSBR multi-protein complexes are mutated in the cerebellar ataxias spinocerebellar ataxia with axonal neuropathy-1, ataxia oculomotor apraxia-1 and ataxia oculomotor apraxia-4. Because XRCC1 is mutated in cerebellar ataxia in this study, it is possible that these complexes could prevent neurodegeneration.
The nuclear enzyme poly (ADP-ribose) polymerase 1 (PARP-1) is the most abundant isoform of the PARP enzyme family. PARP-1 functions as a DNA damage sensor and signaling molecule binding to both single and double-stranded DNA breaks. Poly (ADP-ribosylation) deliberates negative charge to histones leading to electrostatic repulsion among histones and DNA, a process implicated in chromatin remodeling, DNA repair, and transcriptional regulation. Numerous transcription factors, DNA replication factors, and signaling molecules have also been shown to become poly (ADP-ribosylated) by PARP-1. Thus, it is possible that persistent unrepaired SSBs might result in prolonged activity of the SSB sensor protein PARP1.
Elevated ADP-ribose levels were also detected in XRCC1-patient fibroblasts, XRCC1-/- RPE-1 cells and fibroblasts from a patient with ataxia oculomotor apraxia-4 (AOA4) after treatment with CPT, which were all dependent on PARP1 activity. The author detected mouse model in which Xrcc1 was conditionally deleted in brain (Xrcc1 Nes-Cre) and found elevated levels of ADP-ribose in the cerebellum of Xrcc1 Nes-Cre mice, suggesting that the loss of XRCC1 can trigger Parp1 hyperactivation in the brain even at endogenous levels of SSBs. Then the author examines whether PARP1 deletion could rescued the cerebellar ataxia observed in Xrcc1 Nes-Cre mice. And found that Parp1 deletion restores normal ADP-ribosylation levels, interneuron density and reduces cerebellar ataxia in Xrcc1 Nes-Cre mice.
 Fig3. ADP-ribosylation levels measured by immunohistochemistry in cerebellar sections from WT mice or mice deleted of Xrcc1 and/or Parp1.
Fig3. ADP-ribosylation levels measured by immunohistochemistry in cerebellar sections from WT mice or mice deleted of Xrcc1 and/or Parp1.
According to the author, PARP1could be a possible drug target for treating cerebellar ataxias associated with unrepaired SSBs. The development of selective inhibitors of PARP1 that prevent DNA binding by this enzyme may have substantial therapeutic potential.
Reference:
1. Hoch NC, Hanzlikova H, Rulten SL et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature, 2016 Dec 21. doi: 10.1038/nature20790.
2. Pál Pacher1 and Csaba Szabó2, Role of Poly (ADP-ribose) polymerase 1 (PARP-1) in Cardiovascular Diseases: The Therapeutic Potential of PARP Inhibitors, Cardiovasc Drug Rev. 2007; 25(3): 235–260
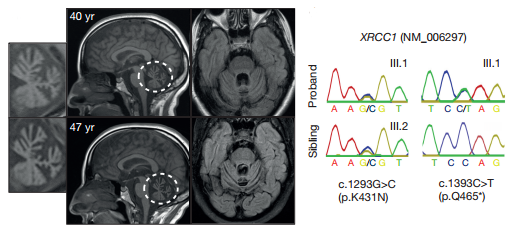 Fig1. XRCC1 mutations are associated with cerebellar ataxia, ocular motor apraxia, and axonal neuropathy
Fig1. XRCC1 mutations are associated with cerebellar ataxia, ocular motor apraxia, and axonal neuropathy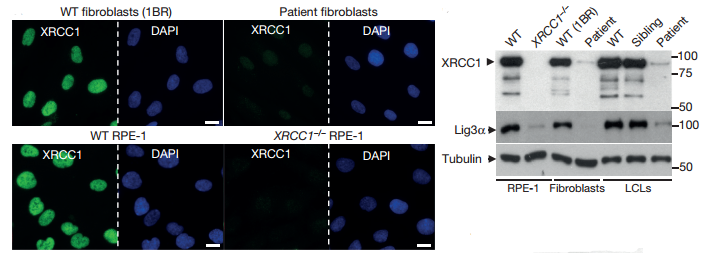 Fig2. Mutations in patient-derived XRCC1 reduce XRCC1 levels
Fig2. Mutations in patient-derived XRCC1 reduce XRCC1 levels Fig3. ADP-ribosylation levels measured by immunohistochemistry in cerebellar sections from WT mice or mice deleted of Xrcc1 and/or Parp1.
Fig3. ADP-ribosylation levels measured by immunohistochemistry in cerebellar sections from WT mice or mice deleted of Xrcc1 and/or Parp1.