
Signal Transduction
- Adaptor Proteins
- Akt Pathway
- Cellular Senescence and Pathways in Aging
- Circadian Rhythm Molecules
- G Protein-Coupled Receptors (GPCRs)
- Intracellular Kinases
- ITIM/ITAM Immunoreceptors and Related Molecules
- Jak/STAT Signaling
- Notch Pathway
- Phosphatases and Regulators
- phospho-Serine/phospho-Threonine Binding Proteins
- Serine Proteases and Regulators
- Ubiquitin and Ubiquitin-like Modifiers
Signal transduction is the process by which cells receive and respond to signals from their environment. These signals can come in various forms, such as hormones, growth factors, neurotransmitters, or light. The process of signal transduction involves a series of sequential steps that convert the extracellular signal into an intracellular response.
1. Signal Reception. The first step in signal transduction is the binding of the signaling molecule, or ligand, to a receptor on the cell surface. This receptor can be an integral membrane protein or a cell surface receptor protein. The binding of the ligand to the receptor triggers a conformational change in the receptor, which then propagates the signal across the cell membrane.
2. Signal Transduction. The next step in signal transduction typically involves the activation of intracellular signaling molecules, often in the form of protein kinases. These kinases phosphorylate target proteins, leading to a cascade of phosphorylation events that amplify and propagate the signal. Additionally, other signaling molecules, such as G proteins or second messengers, may also be involved in the transduction process.
3. Cellular Response. Once the signal has been transmitted intracellularly, it can have a variety of effects on the cell. These effects can include changes in gene expression, alterations in protein activity, cytoskeletal rearrangements, or changes in cellular metabolism. The specific response to a signal depends on the cell type and the nature of the signal itself. This response can vary depending on the specific signaling pathway activated and the target cells involved. It can include changes in cell growth, differentiation, migration, or metabolism, among other cellular functions.
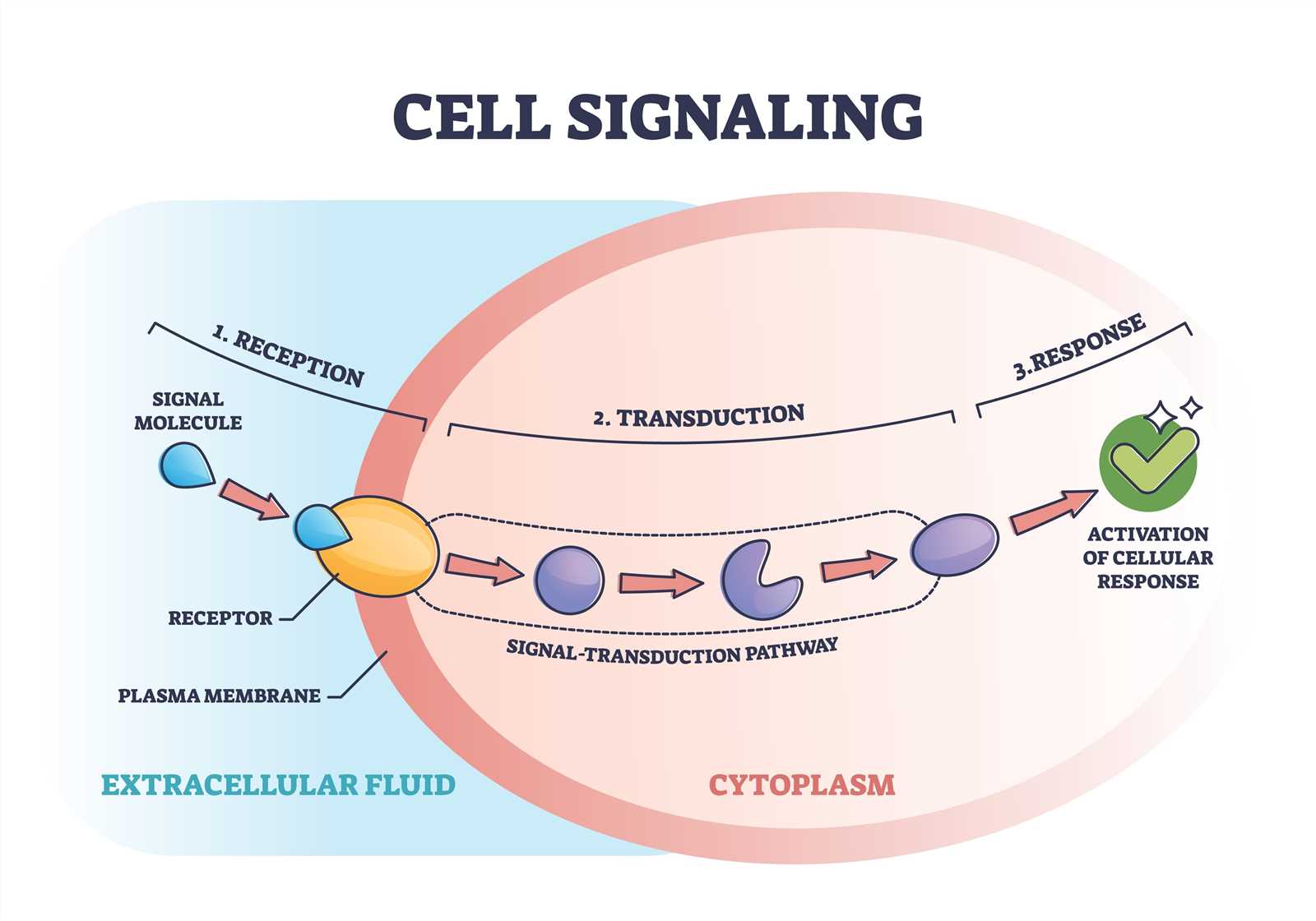
Signal transduction plays a crucial role in cellular communication and is essential for many biological processes, including development, growth, and immune response. Dysregulation of signal transduction pathways can lead to various diseases, such as cancer, diabetes, or neurological disorders.
Overall, signal transduction is a complex and highly regulated process that allows cells to sense and respond to their environment. By understanding the mechanisms underlying signal transduction, researchers hope to gain insights into cellular functions and develop new therpies for diseases.
Significance of the Study of Signal Transduction
The study of signal transduction is significant because it helps us understand how cells receive and process information from their environment, communicate with one another, and ultimately carry out various physiological functions. Some of the key reasons why the study of signal transduction is important include:
1. Cell Communication: Signal transduction allows cells to communicate with each other by transmitting molecular signals, such as hormones or neurotransmitters, from one cell to another. Understanding how these signals are processed and transmitted is crucial for understanding the complex network of interactions that occur within and between cells.
2. Disease Mechanisms: Signal transduction pathways play a central role in the development and progression of various diseases, including cancer, diabetes, and cardiovascular disorders. By unraveling the molecular mechanisms underlying these pathways, researchers can identify potential targets for therapeutic intervention and develop new treatments.
3. Drug Development: Many drugs on the market today target components of signal transduction pathways. Understanding how these pathways function can help identify novel drug targets and improve the design and efficacy of existing therapies.
4. Development and Differentiation: Signal transduction is essential for proper development and differentiation of cells and tissues. Studying how signals are transmitted and interpreted during these processes can provide insights into normal development as well as potential defects that cause developmental disorders.
5. Evolutionary Insights: Signal transduction mechanisms are highly conserved across species, meaning that studying them in model organisms can provide insights into fundamental biological processes that are relevant to humans. Understanding the evolutionary history and function of these pathways can deepen our understanding of biology as a whole.
Research Tools for Signal Transduction
As a leading biotechnology company, Creative BioMart offers a wide range of resources to support research in signal transduction. They provide recombinant proteins, cell and tissue lysates, and protein pre-coupled magnetic beads, all of which are essential tools for studying the components and mechanisms of signal transduction pathways.
In addition to these products, Creative BioMart also offers customized services and other extensive technical and educational resources. These resources aim to assist researchers in their studies by providing comprehensive information and support for various aspects of signal transduction research.
The resources on our website are divided into several categories, each focusing on a specific area of signal transduction research. Categories include:
Adaptor Proteins
Akt Pathway
Cellular Senescence and Pathways in Aging
Circadian Rhythm Molecules
G Protein-Coupled Receptors (GPCRs)
Intracellular Kinases
ITIM/ITAM Immunoreceptors and Related Molecules
Jak/STAT Signaling
Notch Pathway
Phosphatases and Regulators
Phospho-Serine/phospho-Threonine Binding Proteins
Serine Proteases and Regulators
Ubiquitin and Ubiquitin-like Modifiers
Please click to view all related molecules/targets and research reagents.
Our Advantages
We hope we can provide you with valuable insights into research in the signal transduction field, for more information or inquiries, please feel free to contact us .