
Refolding of Inclusion Body Proteins from <em>E. Coli</em>
Expression of proteins in E. coli often leads to the formation of dense insoluble particles, known as inclusion bodies (IBs), containing most of the recombinant proteins in incorrect steric structures without the desired biological activities. How to obtain their native activity and structure, it is the topic we will discuss in this video.
First, of course, we need to understand how protein folding in vivo.
- The E. coli cytoplasm contains a wide variety of different biomolecules, such as lipids, nucleic acids, cytoskeletal components, and other macromolecules, which are sometimes collectively termed crowding agents. An optimal concentration of crowding agents or confinement size exists at which maximum folding yield is obtained.
- Molecular chaperon, which inhibit the generation of non-active conformations and aggregates. One kind chaperones could stabilize nascent and newly synthesized chains and prevent aggregation by forming reversible complexes until the unfolded chains have folded properly. The other kind is large cylindrical chaperonin complexes that provide physically defined compartments (confinement) in which a complete protein, or a protein domain, can fold.
- Protein disulfide isomerase (PDI) catalyzes thiol–disulfide interchange reactions and promotes protein disulfide formation, isomerization, and reduction.
- A quality control system is essential to protein folding in vivo, to ensure the export of native proteins from the endoplasmic reticulum (ER). This system recognizes incorrectly or partially folded proteins and captures these in the ER for subsequent refolding or degradation by the proteasome.
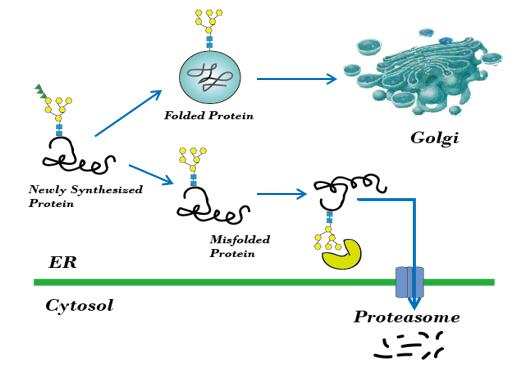 Fig1. ER control system
Fig1. ER control system
In conclusion, the biochemical machinery of protein folding in vivo, which includes the crowding and confinement mechanisms, molecular chaperones and foldases, and the quality control system, has ensured that protein folding in the cell is a highly efficient process. The chemical machinery used for folding in vivo may inspire the design of de novo refolding techniques in vitro, in which the solution environment is tailored for optimal protein folding.
Besides, protein folding also involved free-energy. Protein folding is driven by the free-energy gradient, and folding occurs when the protein attains its thermodynamically most stable form. Protein folding occurs through a myriad of pathways, as illustrated by the ragged-energy funnel; this yields not only the native conformation but also misfolded states and aggregates. Thus, folding is the art of establishing a kinetic partition that drives the protein toward the native conformation. This time we won’t talk further about the energy part.
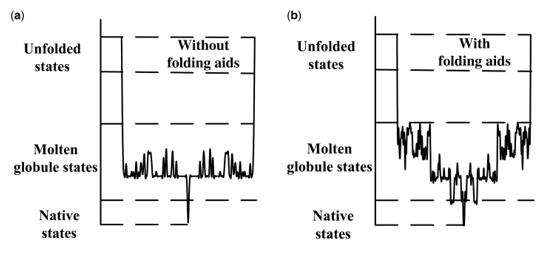 Fig2. Free-energy Gradient
Fig2. Free-energy Gradient
When we express recombinant protein, we prefer soluble status to insoluble (inclusion body, IB), for it is easier to work with a soluble starting material than insoluble IBs. In order to increase soluble proportion, we need to consider expression systems, including a different promoter, using a low copy number plasmid or change host, and culture conditions such as changes in pH, minimal media, concentration of inducer (IPTG), induction temperature, and so on. However, even consider all factors, there are still lots of recombinant protein expressed in IB form. Besides, there is a special benefit for IB production because the IB could be easily separated from large amounts of soluble impurities in the culture by wash and centrifugation. The expression level in IB is usually much higher than in a soluble system. Therefore, IB expression is still widely used in biopharmaceuticals production situations. The development of an efficient refolding process is thus very important for increasing productivity.
Next, we will talking about IB refolding methods, you may concerning most.
First step is releasing IBs from E. coli cells. On the laboratory scale, sonication (ultrasound producing cavitation) is a common method. Others include French press, enzymatic or chemical cleavage of the cells, and freeze–thawing. For larger scale continuous disruption, high pressure homogenization or bead milling are widely used.
Secondly, separated IBs from the cell homogenate after cell disruption to get rid of the cell debris and other soluble impurities. The standard method is differential centrifugation, which often means elaborate washing procedures with a variety of buffer compositions in order to free the IBs from the cell debris and soluble contaminants. Another method is size-exclusion chromatography (SEC) using a gel media with an exclusion limit that allows all cell homogenate components except the IBs to penetrate the gel network.
Third step is IBs solubilization. The vast majority of solubilization procedures rely on strong denaturants such as 6 M guanidine hydrochloride or 8 M urea. If the protein contains disulfide bonds, a reducing agent such as DTT should be added. The example are collected by centrifugation and then dissolved in solubilization buffer, which is ready for further refolding operation.
Fourthly, we need purify IBs before refolding. When IBs have been solubilized, the denatured protein totally reduced, and the solution is free from particulates, initial purification of the random coil target protein is possible using a variety of chromatographic techniques, such as SEC, ion exchange chromatography, or columns with solid-phase.
Finally, the most important step, refolding procedure. There are two problems in refolding process, aggregation and misfolding. Aggregation can be regarded purely as a statistical problem. Thus, the larger the distance between the individual protein molecules in the refolding buffer, the less risk of molecular collision and possible interaction leading to the formation of aggregates. The solution to this problem is simple: reduce the protein concentration in the refolding buffer and you may increase the yield of correctly folded protein. The next problem is misfolding. The refolding process is composed of two stages. The first, mainly based on hydrophobic collapse, is relatively fast and results in the formation of intermediate structures, often called molten globules. These are subsequently, more slowly, reassembled into the final native tertiary structure including reoxidation to form the correct disulfide bridges. Unfortunately, there is no generally applicable solution to this problem. Every protein is unique and thus it is necessary to design individual refolding schemes. However, there are certain rules for reference. If the native protein contains disulfide bridges, it is mandatory to first see that all these are fully reduced. It is then advisable to supply the refolding buffer with a suitable redox system allowing reshuffling of the disulfide bridges, which often facilitates the formation of structurally correct bridges by reoxidation. The more disulfide bridges the protein contains, the more difficult the process and the more important it is to find the optimal redox system.
Here, we will introduce two most used method of IBs refolding.
- Refolding by Dilution
Dilution of the solubilized protein directly into the renaturation buffer is the most commonly used method in small scale refolding studies because of its simplicity. However, protein concentration has to be carefully controlled to prevent aggregation. Also, dilution is time-consuming and buffer-consuming. Thus, for large scale production, dilution may require subsequent concentration after the refolding. A method called “pulse renaturation” has been reported that has given 10% higher yield compared with batch dilution.
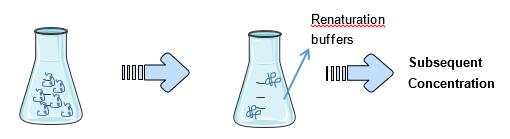 Fig3. Refolding by dilution
Fig3. Refolding by dilution - Refolding by Chromatography
In chromatographic refolding, the solid medium acts as a kind of chaperone or assistant to help the protein refolding occur in a correct way, which minimizes misfolding and aggregation. The feed solution containing denatured protein and denaturant is loaded into the column packed with porous microspheres. Renaturation buffers are introduced to elute the denatured protein to move through the column. During this process, simultaneous refolding and adsorption take place. The solid phase helps the correct folding of the protein. At the column outlet, the protein may exit in the correct form.
Chromatographic refolding is a very complicated process. It is presently difficult to describe the mechanism of chromatographic refolding, because information is very limited. Here, we discuss the molecular action and column control strategy in chromatographic refolding processes.
- Size-exclusion Chromatography (SEC)
The pores of the SEC media have selectivity regarding the size and shape of the proteins. Unfolded protein has a stretched, long shape that is difficult to insert into pores. The correctly formed product, being compact in shape and size, can access the depths of the pores, and is therefore separated from unfolded and partially folded ones. Furthermore a gradient refolding system is used in SEC. It is based on a decreasing urea gradient to provide a gentle and easily controllable environment for protein refolding. In this process a quick change in urea concentration is avoided. The procedure is gentle, providing a slow change of the environment so that the protein refolds gradually.
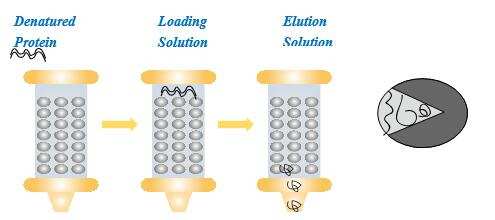 Fig4. SEC refolding mechanism
Fig4. SEC refolding mechanism - Ion-Exchange Chromatography (IEC)
The denatured protein is adsorbed onto the surface of the IEC medium. As the concentration of denaturant in the liquid phase decreases, the protein begins to fold. The folding could take place on the solid surface or away from the surface depending on the protein and its environment. Partial folded protein could also be adsorbed by the solid surface through ionic interaction. The ideal situation is that the protein finishes its refolding process to form a correct structure, and exits the column. Because the adsorption of protein could separate individual molecules, their refoldings do not interfere with one another, and the chance of aggregation is minimized.
However, it may be difficult to optimize denaturant concentration and pH simultaneously in a refolding process, especially in a large scale production. Thus, a dual-gradient IEC process was introduced to enhance the refolding recovery at high protein concentration. At high pH, far away from the protein’s isoelectric point, aggregate formation was prevented, while at low pH near the isoelectric point the establishment of a biologically active conformation was facilitated.
- Hydrophobic-Interaction Chromatography (HIC)
The mechanism involves hydrophobic interaction between immobilized hydrophobic ligands and the proteins in solution. Although most hydrophobic amino acids are buried in the interior of globular proteins, some of them are exposed on the protein surface. These can interact with hydrophobic ligands on the HIC gel. The amount of exposed hydrophobic amino acids differs between proteins and so does the ability of proteins to interact with HIC gels. Considering hydrophobic interactions are the dominant forces in protein folding and structure stabilization, HIC can be an artificial chaperone system through its interaction with the denatured protein. Furthermore, HIC can directly deal with guanidine-HCl denatured proteins, which is an advantage over IEC refolding, which cannot work in high salt solution.
However, successful polypeptide folding is also dependent on undisturbed hydrophobic interaction forces. This is why HIC or RPC interactions should not be so strong as to prevent proper protein refolding.
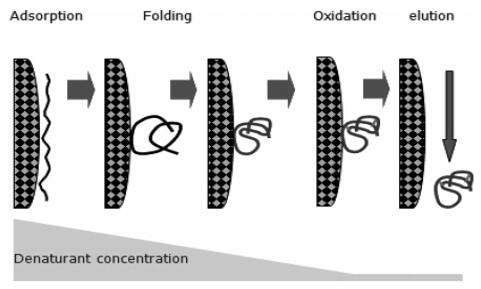 Fig5. IEC and HIC refolding mechanism
Fig5. IEC and HIC refolding mechanism - Other Chromatographic Techniques
Besides the above mentioned techniques, immobilized metal-ion affinity chromatography (IMAC) has opened new prospects for efficient purification and refolding of proteins equipped with engineered polyhistidine tags. Affinity chromatography (AFC) using various ligands can also be used to refold proteins, such as chaperones GroEL and GroES can bind to nascent or unfolded polypeptides and/or their folding intermediates, preventing improper polypeptide chain interactions that lead to aggregation. Other chromatographic such as liposome chromatography, β-Cyclodextrin chromatography and triton X-100 chromatography could also be used for IBs refolding.
- Size-exclusion Chromatography (SEC)
We provide IBs purification services in our company. If you have any questions on protein purification, welcome to contact us for details!