Uncategorized Thursday, 2016/08/04
Ito et al. found that when RIPK1-kinase dependent signaling is disrupted in mice that lack optineurin, necroptosis is inhibited and axonal pathology is reversed. They share their findings on Science Magazine (Science 05 Aug 2016: Vol. 353, Issue 6299, pp. 603-608 DOI: 10.1126/science.aaf6803). We share this articles here for that Creative BioMart can provides kinds of recombinant and native Receptor-interacting kinase 1 (RIPK1) and Optn.
[table caption="Related Products" width="800" colwidth="120|280|150" colalign="left|left|center|center|center"]
Cat. #,Product name,Source(Host),Species,Conjugate
RIPK1-14243M,Recombinant Mouse RIPK1 Protein,Mammalian Cells,Mouse,His
RIPK1-55H,Recombinant Human RIPK1,Sf 9 Insect cells,Human,GST
RIPK1-5972C,Recombinant Chicken RIPK1,Mammalian Cells,Chicken,His
OPTN-12178M,Recombinant Mouse OPTN Protein,Mammalian Cells,Mouse,His
OPTN-197H,Recombinant Human OPTN,E. coli,Human,GST
OPTN-5854Z,Recombinant Zebrafish OPTN,Mammalian Cells,Zebrafish,His
[/table]
Loss-of-function mutations in the optineurin (OPTN) gene have been implicated in both familial and sporadic cases of amyotrophic lateral sclerosis (ALS), a devastating degenerative motor neuron disease. The Optn gene encodes an ubiquitin-binding protein involved in tumor necrosis factor–α (TNFα) signaling but is dispensable for nuclear factor κB (NF-κB) activation. It is still unclear how the loss of function of OPTN leads to human ALS.
Receptor-interacting kinase 1 (RIPK1) is a critical regulator of cell death and inflammation. RIPK1 regulates necroptosis, a form of regulated necrotic cell death, by promoting the sequential activation of two downstream targets, RIPK3 and mixed lineage kinase domain–like protein (MLKL). Application of necrostatin-1 (7-Cl-O-Nec-1) (Nec-1s), a highly specific inhibitor of RIPK1 kinase activity, blocks necroptosis and inflammation in vitro and in vivo. However, the pathophysiological significance of RIPK1 and necroptosis in the genetic context of human diseases remains to be established.
To understand the mechanism by which the loss of OPTN could lead to ALS, we developed Optn–/– mice. We examined the impact of Optn loss in the spinal cord of Optn–/– mice. The number and morphology of spinal cord motor neurons in Optn–/– mice were indistinguishable from wild-type (WT) mice. However, from the age of 3 weeks to 2 years, we observed a marked reduction in the number of motor axons and abnormal myelination in the ventrolateral spinal cord white matter in the Optn–/– mice (Fig. 1, A to D). The axonal pathology presented as a decompaction of myelin sheaths with a decreased g-ratio (axon diameter/axon- plus- myelin diameter), an increased number of large-diameter axons, and a decreased axonal number in the ventrolateral white matter (Fig. 1, B to D), which suggested degeneration and swelling of motor neuron axons in Optn–/– mice. This finding is similar to the axonal pathology observed in the spinal cords of ALS patients in the early stages of the disease. The pathology was progressive—a reduction in axonal numbers was observed at 12 weeks or older but not at 3 weeks. Similar pathology was observed in the ventral roots of motor axons in Optn–/– mice. In addition, denervation of neuromuscular junctions in the tibialis anterior muscle was observed in Optn–/–mice. Thus, OPTN deficiency leads to axonal pathology without affecting motor neuron cell bodies. Consistent with this notion, we observed a significant increase in the number of cells positive for terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL+ cells) in the ventrolateral white matter of spinal cords of Optn–/– mice (Fig. 1E). Thus, Optn deficiency sensitizes cells to cell death in the spinal cord white matter of Optn–/– mice.
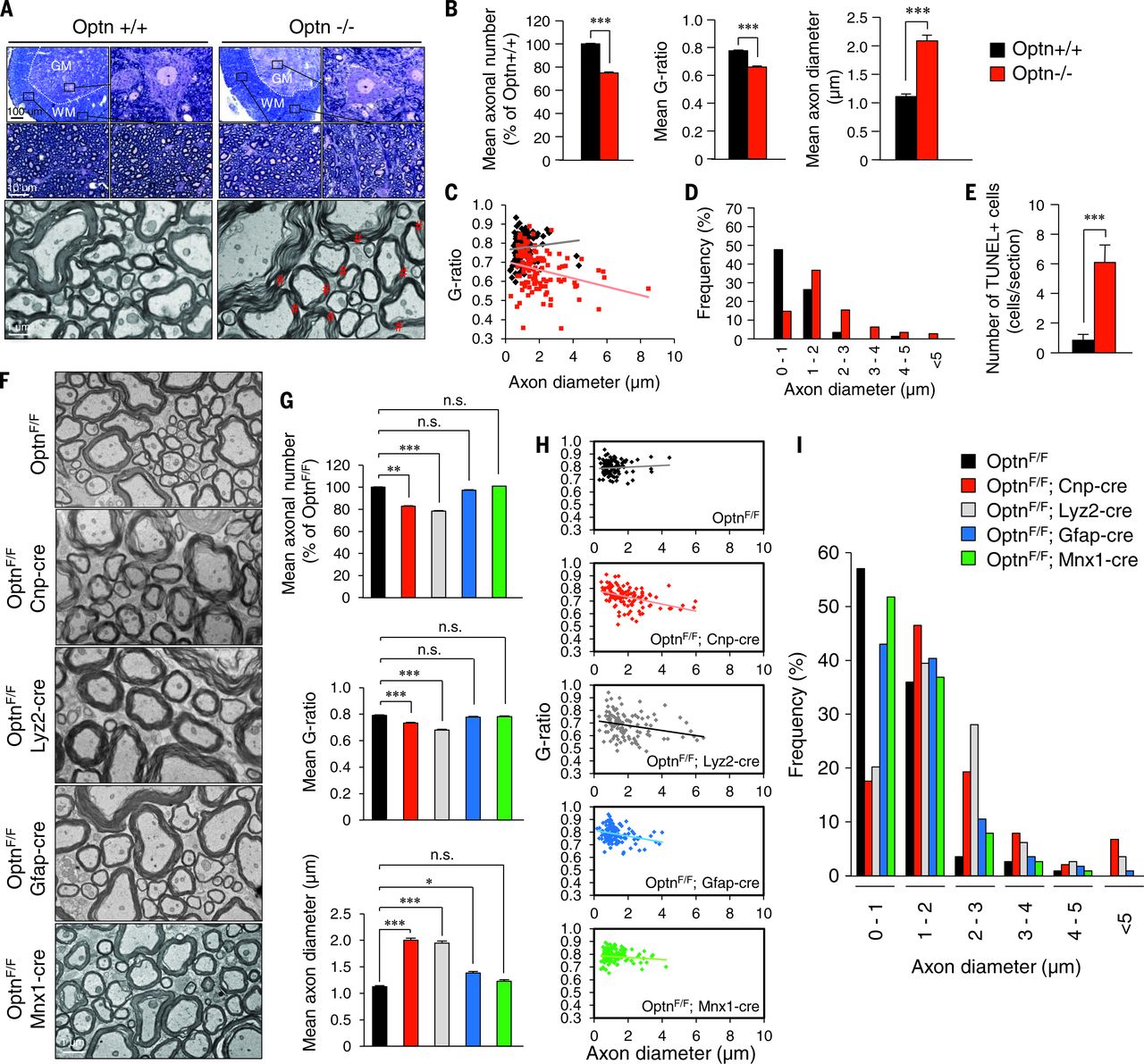 Fig. 1 Optn deficiency in oligodendrocyte and myeloid lineages promotes axonal loss and dysmyelination in the spinal cords of Optn–/– mice.
To determine the cell types involved in mediating Optn deficiency–induced axonal degeneration, we generated-linage specific deletion of Optn using Cnp-cre, Lyz2-cre, Gfap-cre, and Mnx1-cre mice. Loss of Optn from oligodendrocytes and myeloid cells, but not from astrocytes or motor neurons, was sufficient to reproduce axonal myelination pathology (Fig. 1, F to I). Furthermore, we induced Optn loss from the microglial lineage by dosing OptnF/F;Cx3cr1Cre mice with tamoxifen for 1 month and also found axonal pathology like that in Optn–/– mice.
We found that knockdown of Optn sensitized cells to necroptosis in our genome-wide small interfering RNA screen (Z-score = −2.07). We further confirmed that knockdown of Optn sensitized L929 cells to necroptosis induced by TNF± or zVAD.fmk. zVAD-induced necrosis is known to involve autocrine TNFα activity. Thus, Optn deficiency sensitized cells to necroptosis. The biochemical hallmarks of necroptosis—including the upshifts of Ripk1, Ripk3, and phospho-MLKL (p-MLKL), as well as the levels of complex IIb—were significantly higher in Optn–/– mouse embryo fibroblasts (MEFs) than in Optn+/+ MEFs stimulated by TNFα, zVAD, or cycloheximide. Note that Optn–/– oligodendrocytes were sensitized to die by TNFα-induced necroptosis but were protected by Nec-1s and in Optn–/–;Ripk1D138N/D138N and Optn–/–;Ripk3–/–double mutants (Fig. 2A). Thus, Optn deficiency can promote necroptosis of oligodendrocytes.
Fig. 1 Optn deficiency in oligodendrocyte and myeloid lineages promotes axonal loss and dysmyelination in the spinal cords of Optn–/– mice.
To determine the cell types involved in mediating Optn deficiency–induced axonal degeneration, we generated-linage specific deletion of Optn using Cnp-cre, Lyz2-cre, Gfap-cre, and Mnx1-cre mice. Loss of Optn from oligodendrocytes and myeloid cells, but not from astrocytes or motor neurons, was sufficient to reproduce axonal myelination pathology (Fig. 1, F to I). Furthermore, we induced Optn loss from the microglial lineage by dosing OptnF/F;Cx3cr1Cre mice with tamoxifen for 1 month and also found axonal pathology like that in Optn–/– mice.
We found that knockdown of Optn sensitized cells to necroptosis in our genome-wide small interfering RNA screen (Z-score = −2.07). We further confirmed that knockdown of Optn sensitized L929 cells to necroptosis induced by TNF± or zVAD.fmk. zVAD-induced necrosis is known to involve autocrine TNFα activity. Thus, Optn deficiency sensitized cells to necroptosis. The biochemical hallmarks of necroptosis—including the upshifts of Ripk1, Ripk3, and phospho-MLKL (p-MLKL), as well as the levels of complex IIb—were significantly higher in Optn–/– mouse embryo fibroblasts (MEFs) than in Optn+/+ MEFs stimulated by TNFα, zVAD, or cycloheximide. Note that Optn–/– oligodendrocytes were sensitized to die by TNFα-induced necroptosis but were protected by Nec-1s and in Optn–/–;Ripk1D138N/D138N and Optn–/–;Ripk3–/–double mutants (Fig. 2A). Thus, Optn deficiency can promote necroptosis of oligodendrocytes.
 Fig. 2 Optn deficiency sensitizes to necroptosis.
The expression levels of Ripk1, Ripk3, and MLKL—the key mediators of necroptosis—were all increased in the spinal cords of Optn–/– mice (Fig. 2B). Furthermore, we detected the interaction of Optn and Ripk1 in spinal cords from WT mice (Fig. 2C). Compared with WT mice, RIPK1 lysine 48 (K48) ubiquitination levels were decreased, whereas Ripk1 mRNA was unchanged in the spinal cords of Optn–/– mice (Fig. 2, D and E). Furthermore, Ripk1 was degraded more slowly in Optn–/– MEFs than that in WT cells (Fig. 2F). Thus, OPTN might control sensitivity to necroptosis by regulating proteasomal turnover of RIPK1.
Phospho-Ser14/15, a marker of Ripk1 activation, was increased in Optn–/– microglia relative to WT microglia, which were inhibited by Nec-1s and Ripk1D138N/D138N mutation (Fig. 2G). Because microglia express little MLKL, we hypothesize that Ripk1 activation in microglia promotes inflammatory signaling not necroptosis. Consistent with this notion, we detected an increased production of multiple proinflammatory cytokines—including interleukins IL-1α, IL-1β, IL-2, and IL-12; interferon-γ (IFN–γ); and TNFα in the spinal cords of Optn–/– mice—which were markedly reduced in the Optn–/–;Ripk1D138N/D138N mice (Fig. 2H). In addition, Optn–/– microglia had elevated TNFα, which was inhibited by Nec-1s. As predicted, the levels of TNFα were also increased in the spinal cords of OptnF/F;Lyz2-cre mice.
To explore the effect of Optn deficiency on transcriptions, we performed RNA sequencing on WT, Optn–/–, and Optn–/–;Ripk1D138N/D138N primary microglia. Co-expression analysis identified a module with ~1300 genes (ME1) differentially expressed between WT and Optn–/–microglia and suppressed by Ripk1D138N/D138N. The top 71 genes in this module include CD14 and CD86, biomarkers for the proinflammatory M1-like state (Fig. 2I). Elevated CD14 and CD86 in Optn–/– microglia were suppressed by Nec-1s and Ripk1D138N/D138N. Thus, Optn deficiency promotes an M1-like inflammatory microglia.
We analyzed the genes differentially expressed in Optn–/– microglia using MSigDB (Molecular Signatures Database) to identify transcription factors with targets that were overrepresented. We found a significant overrepresentation of the predicted Sp1 transcription factor targets in the ME1 module with a network of 225 Sp1 targets regulated by RIPK1. Increased production of TNFα and the death of L929 cells were blocked by knockdown of Sp1 and by Nec-1s. Thus, loss of Optn in the spinal cord may increase RIPK1-dependent inflammation.
We examined the involvement of necroptosis in Optn–/– mice in vivo. The increase in TUNEL+ cells and the axonal pathology of Optn–/– mice were all rescued in the Optn–/–;Ripk1D138N/D138N double-mutant and the Optn–/–;Ripk3–/– double-mutant mice and by Nec-1s (Fig. 3, A to E). Behaviorally, Optn–/– mice showed no difference in total locomotor activity, whereas the vertical rearing activity was significantly reduced compared with that of WT mice (Fig. 3, F to H). Thus, Optn deficiency leads to hindlimb weakness. Furthermore, the vertical rearing deficit in Optn–/– mice was rescued pharmacologically by Nec-1s and genetically in the Optn–/–;Ripk1D138N/D138N mice and Optn–/–;Ripk3–/– mice. Thus, Optn deficiency leads to the activation of necroptotic machinery to promote axonal pathology.
Fig. 2 Optn deficiency sensitizes to necroptosis.
The expression levels of Ripk1, Ripk3, and MLKL—the key mediators of necroptosis—were all increased in the spinal cords of Optn–/– mice (Fig. 2B). Furthermore, we detected the interaction of Optn and Ripk1 in spinal cords from WT mice (Fig. 2C). Compared with WT mice, RIPK1 lysine 48 (K48) ubiquitination levels were decreased, whereas Ripk1 mRNA was unchanged in the spinal cords of Optn–/– mice (Fig. 2, D and E). Furthermore, Ripk1 was degraded more slowly in Optn–/– MEFs than that in WT cells (Fig. 2F). Thus, OPTN might control sensitivity to necroptosis by regulating proteasomal turnover of RIPK1.
Phospho-Ser14/15, a marker of Ripk1 activation, was increased in Optn–/– microglia relative to WT microglia, which were inhibited by Nec-1s and Ripk1D138N/D138N mutation (Fig. 2G). Because microglia express little MLKL, we hypothesize that Ripk1 activation in microglia promotes inflammatory signaling not necroptosis. Consistent with this notion, we detected an increased production of multiple proinflammatory cytokines—including interleukins IL-1α, IL-1β, IL-2, and IL-12; interferon-γ (IFN–γ); and TNFα in the spinal cords of Optn–/– mice—which were markedly reduced in the Optn–/–;Ripk1D138N/D138N mice (Fig. 2H). In addition, Optn–/– microglia had elevated TNFα, which was inhibited by Nec-1s. As predicted, the levels of TNFα were also increased in the spinal cords of OptnF/F;Lyz2-cre mice.
To explore the effect of Optn deficiency on transcriptions, we performed RNA sequencing on WT, Optn–/–, and Optn–/–;Ripk1D138N/D138N primary microglia. Co-expression analysis identified a module with ~1300 genes (ME1) differentially expressed between WT and Optn–/–microglia and suppressed by Ripk1D138N/D138N. The top 71 genes in this module include CD14 and CD86, biomarkers for the proinflammatory M1-like state (Fig. 2I). Elevated CD14 and CD86 in Optn–/– microglia were suppressed by Nec-1s and Ripk1D138N/D138N. Thus, Optn deficiency promotes an M1-like inflammatory microglia.
We analyzed the genes differentially expressed in Optn–/– microglia using MSigDB (Molecular Signatures Database) to identify transcription factors with targets that were overrepresented. We found a significant overrepresentation of the predicted Sp1 transcription factor targets in the ME1 module with a network of 225 Sp1 targets regulated by RIPK1. Increased production of TNFα and the death of L929 cells were blocked by knockdown of Sp1 and by Nec-1s. Thus, loss of Optn in the spinal cord may increase RIPK1-dependent inflammation.
We examined the involvement of necroptosis in Optn–/– mice in vivo. The increase in TUNEL+ cells and the axonal pathology of Optn–/– mice were all rescued in the Optn–/–;Ripk1D138N/D138N double-mutant and the Optn–/–;Ripk3–/– double-mutant mice and by Nec-1s (Fig. 3, A to E). Behaviorally, Optn–/– mice showed no difference in total locomotor activity, whereas the vertical rearing activity was significantly reduced compared with that of WT mice (Fig. 3, F to H). Thus, Optn deficiency leads to hindlimb weakness. Furthermore, the vertical rearing deficit in Optn–/– mice was rescued pharmacologically by Nec-1s and genetically in the Optn–/–;Ripk1D138N/D138N mice and Optn–/–;Ripk3–/– mice. Thus, Optn deficiency leads to the activation of necroptotic machinery to promote axonal pathology.
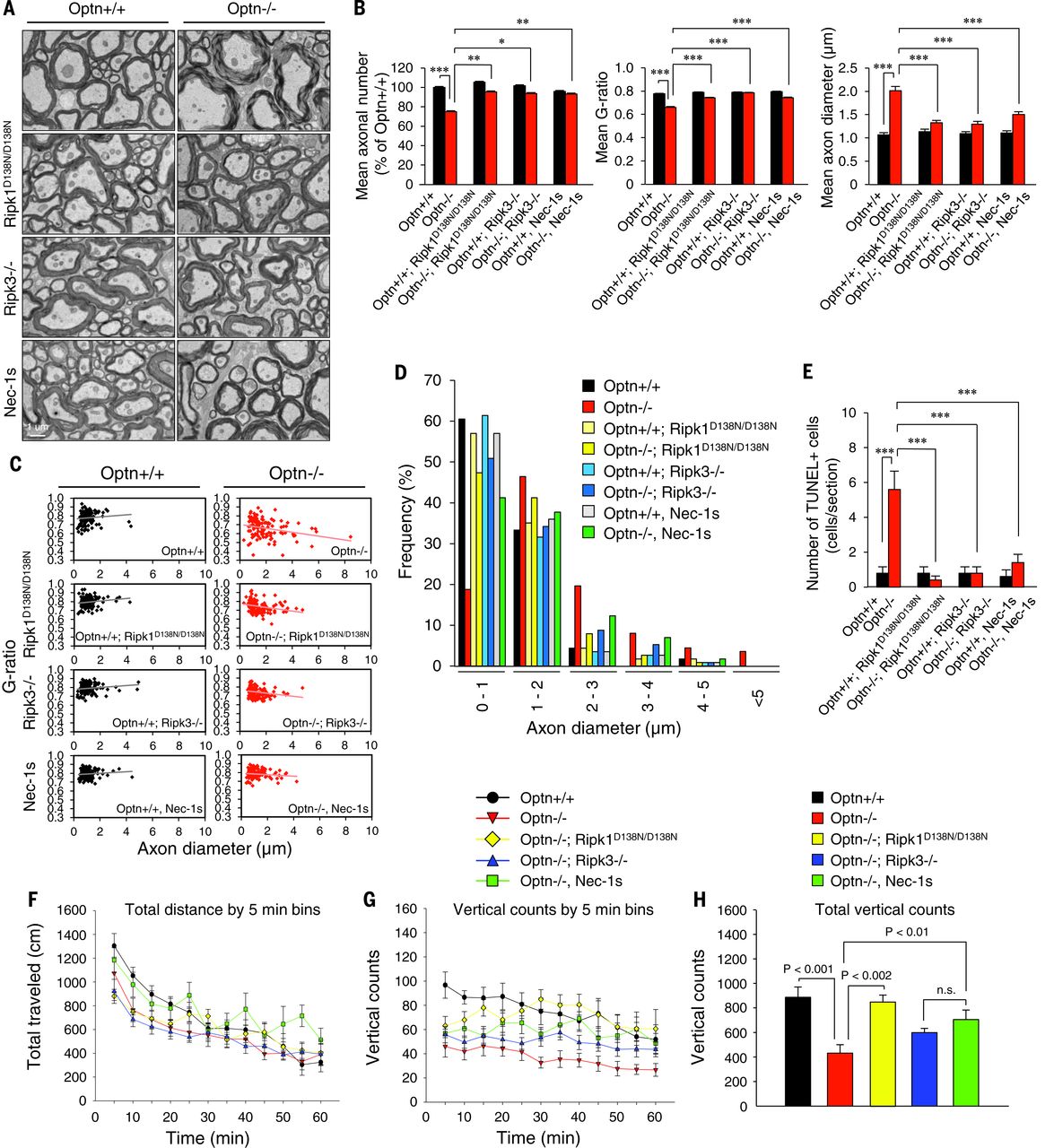 Fig. 3 Ripk1 and Ripk3 mediate axonal pathology in the spinal cords of Optn–/–mice.
To explore the involvement of RIPK1-mediated axonal pathology in ALS in general, we used SOD1G93A transgenic mice. Oligodendrocytes in SOD1G93A mice degenerate early, but the mechanism is unclear. We found that the expression of Ripk1, Ripk3, and MLKL in the spinal cords of SOD1G93A transgenic mice was elevated (Fig. 4A). In addition, we observed a similar axonal pathology as that of Optn–/– mice in SOD1G93A mice before the onset of motor dysfunction (Fig. 4, B and C). Furthermore, these axonal myelination defects were blocked and motor dysfunction onset was delayed genetically by Ripk3 knockout or by oral administration of Nec-1s (Fig. 4, D and E). Thus, although we cannot rule out the contribution of Ripk1 or other proapoptotic factors to the degeneration of motor neuron cell bodies, the activation of necroptosis contributes to axonal pathology and motor dysfunction in the SOD1G93A transgenic mice.
Fig. 3 Ripk1 and Ripk3 mediate axonal pathology in the spinal cords of Optn–/–mice.
To explore the involvement of RIPK1-mediated axonal pathology in ALS in general, we used SOD1G93A transgenic mice. Oligodendrocytes in SOD1G93A mice degenerate early, but the mechanism is unclear. We found that the expression of Ripk1, Ripk3, and MLKL in the spinal cords of SOD1G93A transgenic mice was elevated (Fig. 4A). In addition, we observed a similar axonal pathology as that of Optn–/– mice in SOD1G93A mice before the onset of motor dysfunction (Fig. 4, B and C). Furthermore, these axonal myelination defects were blocked and motor dysfunction onset was delayed genetically by Ripk3 knockout or by oral administration of Nec-1s (Fig. 4, D and E). Thus, although we cannot rule out the contribution of Ripk1 or other proapoptotic factors to the degeneration of motor neuron cell bodies, the activation of necroptosis contributes to axonal pathology and motor dysfunction in the SOD1G93A transgenic mice.
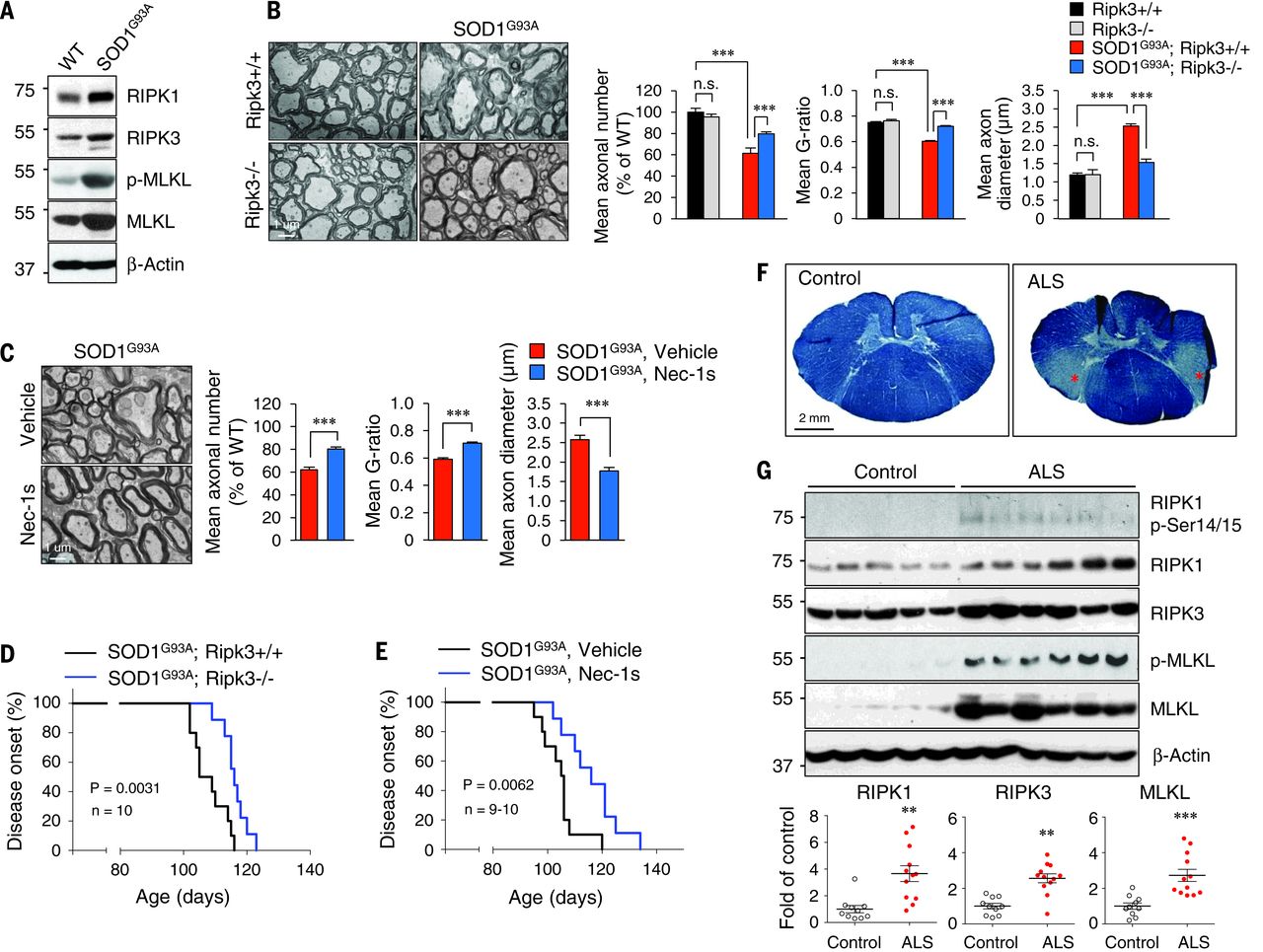 Fig. 4 RIPK1- and RIPK3- mediated axonal pathology is a common mechanism in ALS.
We next characterized the role of RIPK1 and necroptosis in human ALS. We found evidence of demyelination in the lateral column white matter of lower spinal cord pathological samples from ALS patients as reported (Fig. 4F). In human ALS pathological samples, we also detected multiple biochemical hallmarks of necroptosis, including increased levels of RIPK1, RIPK3, and MLKL and increased RIPK1 p-Ser14/15 and p-MLKL in both microglia and oligodendrocytes (Fig. 4G). Note that p-MLKL was primarily localized in the white matter, where demyelination was found.
Taken together, our results provide a direct connection between Wallerian-like degeneration induced by OPTN deficiency and RIPK1-regulated necroptosis and inflammation. By promoting both inflammation and cell death, RIPK1 may be a common mediator of axonal pathology in ALS. Because RIPK1 is recruited specifically to the TNF receptor TNFR1 to mediate the deleterious effect of TNFα, blocking RIPK1 may provide a therapeutic option for the treatment of ALS without affecting TNFR2. Finally, given the recruitment of OPTN to intracellular protein aggregates found in pathological samples from patients with Alzheimer’s disease, Parkinson’s disease, Creutzfeldt-Jakob disease, multiple system atrophy, and Pick’s disease, a possible role of RIPK1 in mediating the wide presence of axonal degeneration in different neurodegenerative diseases should be considered.
Reference
You can visit http://science.sciencemag.org/content/353/6299/603 for more details.
Fig. 4 RIPK1- and RIPK3- mediated axonal pathology is a common mechanism in ALS.
We next characterized the role of RIPK1 and necroptosis in human ALS. We found evidence of demyelination in the lateral column white matter of lower spinal cord pathological samples from ALS patients as reported (Fig. 4F). In human ALS pathological samples, we also detected multiple biochemical hallmarks of necroptosis, including increased levels of RIPK1, RIPK3, and MLKL and increased RIPK1 p-Ser14/15 and p-MLKL in both microglia and oligodendrocytes (Fig. 4G). Note that p-MLKL was primarily localized in the white matter, where demyelination was found.
Taken together, our results provide a direct connection between Wallerian-like degeneration induced by OPTN deficiency and RIPK1-regulated necroptosis and inflammation. By promoting both inflammation and cell death, RIPK1 may be a common mediator of axonal pathology in ALS. Because RIPK1 is recruited specifically to the TNF receptor TNFR1 to mediate the deleterious effect of TNFα, blocking RIPK1 may provide a therapeutic option for the treatment of ALS without affecting TNFR2. Finally, given the recruitment of OPTN to intracellular protein aggregates found in pathological samples from patients with Alzheimer’s disease, Parkinson’s disease, Creutzfeldt-Jakob disease, multiple system atrophy, and Pick’s disease, a possible role of RIPK1 in mediating the wide presence of axonal degeneration in different neurodegenerative diseases should be considered.
Reference
You can visit http://science.sciencemag.org/content/353/6299/603 for more details.