Uncategorized Saturday, 2017/04/08
Body weight is determined by energy balance. When the energy intake equals energy expenditure, the body weight remains unchanged. On the other hand, the body weight grow when the energy surplus. Energy balance is critical for survival and health, and control of food intake is an integral part of this process. Excess energy intake has become a scapegoat for the obesity epidemic in developed countries. While it is responsible in a large part due to unlimited access to food, peoples’ genes play a significant role as well. Central signals as well as several hormones and circulating peptides influence food intake and/or energy expenditure in a coordinated manner to regulate body weight. Appetite is regulated by signals at three levels in the human body: cellular energy sensors, peripheral signals and the central nervous system (CNS). The major cellular energy sensor is the protein AMPK (AMP-activated protein kinase). The peripheral system generates and relays “hunger” or “satiety” signals to the CNS and the CNS makes final decisions.
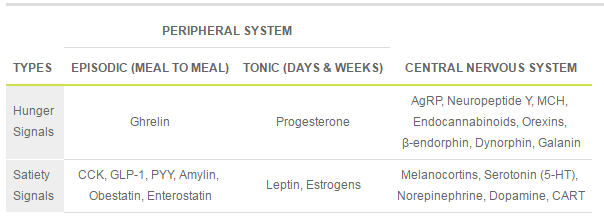
Table1. Hunger and Satiety Signals
 [caption id="attachment_605" align="aligncenter" width="411"]
[caption id="attachment_605" align="aligncenter" width="411"]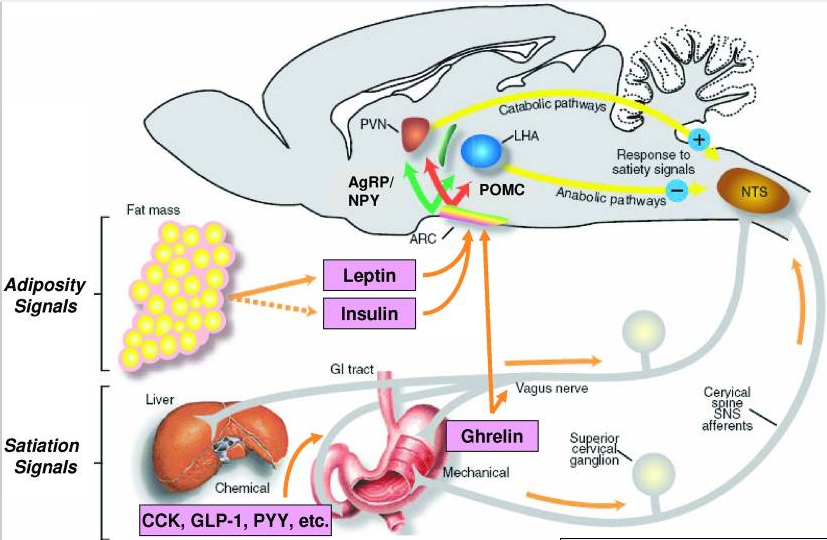 Fig1. Hunger and Satiety Signals Influence Appetite and Body Weight[/caption]
Besides hormones and peptides cited above, there are other signals could influence food intake and energy expenditure.
Lipocalin-2 (LCN-2), 25 kDa, secreted glycoprotein, a member of the lipocalin superfamily, is released from the adipocytes, neutrophils, macrophages, hepatocytes, and epithelial cells. LCN-2 expression in adipose tissue is elevated in obesity. The first evidence for the increased expression of LCN2 in obesity came from a study by Soukas et al., in which they observed the increased expression of LCN2 in white adipose tissue (WAT) of ob/ob mice. Experimental studies have demonstrated the role of LCN-2 in glucose metabolism, lipid metabolism, insulin resistance, and inflammation.
At cell and molecular level, obesity reflects a state of low-grade chronic inflammation. Metabolic inflammation is characterized by the dysregulation of cytokine and adipocytokine expression in adipose tissue. LCN2 might have a major role in intestinal inflammation, and it is increasingly recognized that dysbiosis and changes in intestinal permeability might contribute to systemic inflammation, insulin resistance, and associated features.
But in a recent research in Nature, the author finds that LCN2, which was previously thought to be exclusively secreted by adipose tissue (an adipokine), is expressed by osteoblasts, at levels that are at least tenfold higher in osteoblasts than in white adipose tissue or other organs.
Mice that lacked LCN2 in osteoblasts (Lcn2osb-/-) had compromised glucose metabolism, as was shown by decreased glucose tolerance, insulin sensitivity, a lack of insulin secretion after glucose or arginine challenge. Lcn2osb-/- mice also showed increased fat weight, total fat mass and body weight. However, none of these parameters were affected in Lcn2fat-/- mice (lacked LCN2 in adipocytes). After refeeding 1-3h, LCN2 level in serum increase threefold, owing to LCN producted by osteoblasts, as it correlated with a 1.6-fold increase in Lcn2 expression in bone, but not in fat or other tissues.
[caption id="attachment_606" align="aligncenter" width="717"]
Fig1. Hunger and Satiety Signals Influence Appetite and Body Weight[/caption]
Besides hormones and peptides cited above, there are other signals could influence food intake and energy expenditure.
Lipocalin-2 (LCN-2), 25 kDa, secreted glycoprotein, a member of the lipocalin superfamily, is released from the adipocytes, neutrophils, macrophages, hepatocytes, and epithelial cells. LCN-2 expression in adipose tissue is elevated in obesity. The first evidence for the increased expression of LCN2 in obesity came from a study by Soukas et al., in which they observed the increased expression of LCN2 in white adipose tissue (WAT) of ob/ob mice. Experimental studies have demonstrated the role of LCN-2 in glucose metabolism, lipid metabolism, insulin resistance, and inflammation.
At cell and molecular level, obesity reflects a state of low-grade chronic inflammation. Metabolic inflammation is characterized by the dysregulation of cytokine and adipocytokine expression in adipose tissue. LCN2 might have a major role in intestinal inflammation, and it is increasingly recognized that dysbiosis and changes in intestinal permeability might contribute to systemic inflammation, insulin resistance, and associated features.
But in a recent research in Nature, the author finds that LCN2, which was previously thought to be exclusively secreted by adipose tissue (an adipokine), is expressed by osteoblasts, at levels that are at least tenfold higher in osteoblasts than in white adipose tissue or other organs.
Mice that lacked LCN2 in osteoblasts (Lcn2osb-/-) had compromised glucose metabolism, as was shown by decreased glucose tolerance, insulin sensitivity, a lack of insulin secretion after glucose or arginine challenge. Lcn2osb-/- mice also showed increased fat weight, total fat mass and body weight. However, none of these parameters were affected in Lcn2fat-/- mice (lacked LCN2 in adipocytes). After refeeding 1-3h, LCN2 level in serum increase threefold, owing to LCN producted by osteoblasts, as it correlated with a 1.6-fold increase in Lcn2 expression in bone, but not in fat or other tissues.
[caption id="attachment_606" align="aligncenter" width="717"]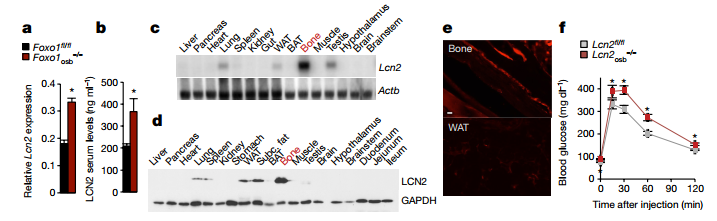 Fig2. LCN2 regulates glucose homeostasis through its expression in osteoblasts.[/caption]
The author finds that LCN2 suppresses appetite by signalling directly in the brain on the hypothalamus. Further investigating the mechanism of LCN2 suppresses food intake, the author finds that only the expression of downstream effectors of MC4R signaling was altered among the pathways which could affect appetite on hypothalamic. In 2008, MC4R mutations were reported to be associated with inherited human obesity. The melanocortin-4 receptor (MC4R) is a G protein-coupled receptor expressed in the brain, where it controls energy balance through pathways including α-melanocyte-stimulating hormone (α-MSH)-dependent signaling.
Besides, the author also finds that LCN2 does not induce phosphorylation of AMPK, ERK1, ERK2 or tyrosine kinase but activated cAMP as efficiently as α-MSH. Secondly, silencing Mc4R or pharmacologically inhibiting its activity abrogated LCN2-induced cAMP activity. Third, LCN2 activated MC4R signaling. Fourth, biotinylated LCN2, at a physiological dose in the hypothalamus, bound to PVH neurons and neurons in the ventromedial nucleus of the hypothalamus (VMH) of Lcn2-/- mice, where MC4R is expressed. Therefore, the author concluded that LCN2 binds to MC4R-expressing hypothalamic neurons and activates MC4R signalling.
There is another question for the conclusion. Does the MC4R necessary for LCN2 activity? The answer is YES! The author found that when treated Mc4r-/- and wild-type mice with LCN2, LCN2 suppressed appetite and decreased body weight in wild-type but not in Mc4r-/- mice. Lcn2osb+/-::Mc4r+/- mice showed a significant increase in appetite, body weight and fat mass, and decreased glucose tolerance and insulin sensitivity compared to Lcn2osb+/- and Mc4r+/- mice. Besides, analysis of patients with MC4R mutations and normal MC4R shows that LCN2 levels were two–fourfold increased patient with MC4R mutations, which suggested that LCN2 regulates MC4R signaling in humans.
This is article concluded that osteoblast-derived LCN2 crosses the blood–brain barrier, and binds to and activates MC4R in PVN neurons of the hypothalamus with potency similar to that of leptin and comparable to the potent α-MSH analogue, MT-II. In addition to appetite-suppressing activities, LCN2 regulates insulin secretion and increases insulin sensitivity and glucose tolerance. Although, it is still unknown for why bone would suppress food intake, this finding provides a new sight for health-keepers. You should also pay attention to your skeleton when you control your appetite and take exercise.
Fig2. LCN2 regulates glucose homeostasis through its expression in osteoblasts.[/caption]
The author finds that LCN2 suppresses appetite by signalling directly in the brain on the hypothalamus. Further investigating the mechanism of LCN2 suppresses food intake, the author finds that only the expression of downstream effectors of MC4R signaling was altered among the pathways which could affect appetite on hypothalamic. In 2008, MC4R mutations were reported to be associated with inherited human obesity. The melanocortin-4 receptor (MC4R) is a G protein-coupled receptor expressed in the brain, where it controls energy balance through pathways including α-melanocyte-stimulating hormone (α-MSH)-dependent signaling.
Besides, the author also finds that LCN2 does not induce phosphorylation of AMPK, ERK1, ERK2 or tyrosine kinase but activated cAMP as efficiently as α-MSH. Secondly, silencing Mc4R or pharmacologically inhibiting its activity abrogated LCN2-induced cAMP activity. Third, LCN2 activated MC4R signaling. Fourth, biotinylated LCN2, at a physiological dose in the hypothalamus, bound to PVH neurons and neurons in the ventromedial nucleus of the hypothalamus (VMH) of Lcn2-/- mice, where MC4R is expressed. Therefore, the author concluded that LCN2 binds to MC4R-expressing hypothalamic neurons and activates MC4R signalling.
There is another question for the conclusion. Does the MC4R necessary for LCN2 activity? The answer is YES! The author found that when treated Mc4r-/- and wild-type mice with LCN2, LCN2 suppressed appetite and decreased body weight in wild-type but not in Mc4r-/- mice. Lcn2osb+/-::Mc4r+/- mice showed a significant increase in appetite, body weight and fat mass, and decreased glucose tolerance and insulin sensitivity compared to Lcn2osb+/- and Mc4r+/- mice. Besides, analysis of patients with MC4R mutations and normal MC4R shows that LCN2 levels were two–fourfold increased patient with MC4R mutations, which suggested that LCN2 regulates MC4R signaling in humans.
This is article concluded that osteoblast-derived LCN2 crosses the blood–brain barrier, and binds to and activates MC4R in PVN neurons of the hypothalamus with potency similar to that of leptin and comparable to the potent α-MSH analogue, MT-II. In addition to appetite-suppressing activities, LCN2 regulates insulin secretion and increases insulin sensitivity and glucose tolerance. Although, it is still unknown for why bone would suppress food intake, this finding provides a new sight for health-keepers. You should also pay attention to your skeleton when you control your appetite and take exercise.