Uncategorized Thursday, 2016/07/21
R. Dixon Dorand et al. have found that Cdk5 disruption can help outcome tumor medulloblastoma. They published their research results in Science Magazine (Science 22 Jul 2016: Vol. 353, Issue 6297, pp. 399-403 DOI: 10.1126/science.aae0477), and we share this wonderful article here. By the way, Creative Biomart provides kinds of molecular tools to promote your research about tumor therapy.
[table caption="Related Products" width="800" colwidth="80|180|180" colalign="left|left|center|center|center"]
Cat. #,Product name,Source(Host),Species,Conjugate
CDK5-3204M,Recombinant Mouse CDK5 Protein,Mammalian Cells,Mouse,His
CDK5-1305R,Recombinant Rat CDK5 Protein,Mammalian Cells,Rat,His
CDK5-186H,Recombinant Human CDK5,Mammalian Cells,Human,His
Cd274-721M,Recombinant Mouse Cd274 Fc-tagged Biotinylated,Human cells,Mouse,Fc
CD274-592H,Recombinant Human CD274 His tagged,HEK293,Human,His/
[/table]
Cyclin-dependent kinase 5 (Cdk5) is a nonstereotypical Cdk whose activity depends on coactivators, p35 and/or p39. A proline-directed serine-threonine kinase, Cdk5 is essential in central nervous system (CNS) development. Cdk5 also contributes to angiogenesis, apoptosis, myogenesis, vesicular transport, and senescence in nonneuronal cells, including tumors, which makes Cdk5 a potential therapeutic target in cancers. We explored whether Cdk5 plays a role in medulloblastoma (MB), a common malignant pediatric CNS tumor.
MB cell lines and clinical specimens expressed Cdk5, p35, and p39 (Fig. 1A). Cdk5-specific kinase activity could be abolished in vitro by roscovitine, a nonselective inhibitor against Cdks 1, 2, 5, 7, and 9. To interrogate Cdk5-specific functions, we disrupted Cdk5 in wild-type murine MB cells (MM1 WT) by short hairpin–mediated RNA interference (MM1 shCdk5) and clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9-targeted mutation (MM1 crCdk5 ), with nontargeting constructs as controls (MM1 shNS and MM1 crNeg). A reduction in Cdk5 was confirmed at the transcript and protein levels. In vitro, there were no significant differences in cell proliferation among all constructs.
[caption id="attachment_515" align="aligncenter" width="300"]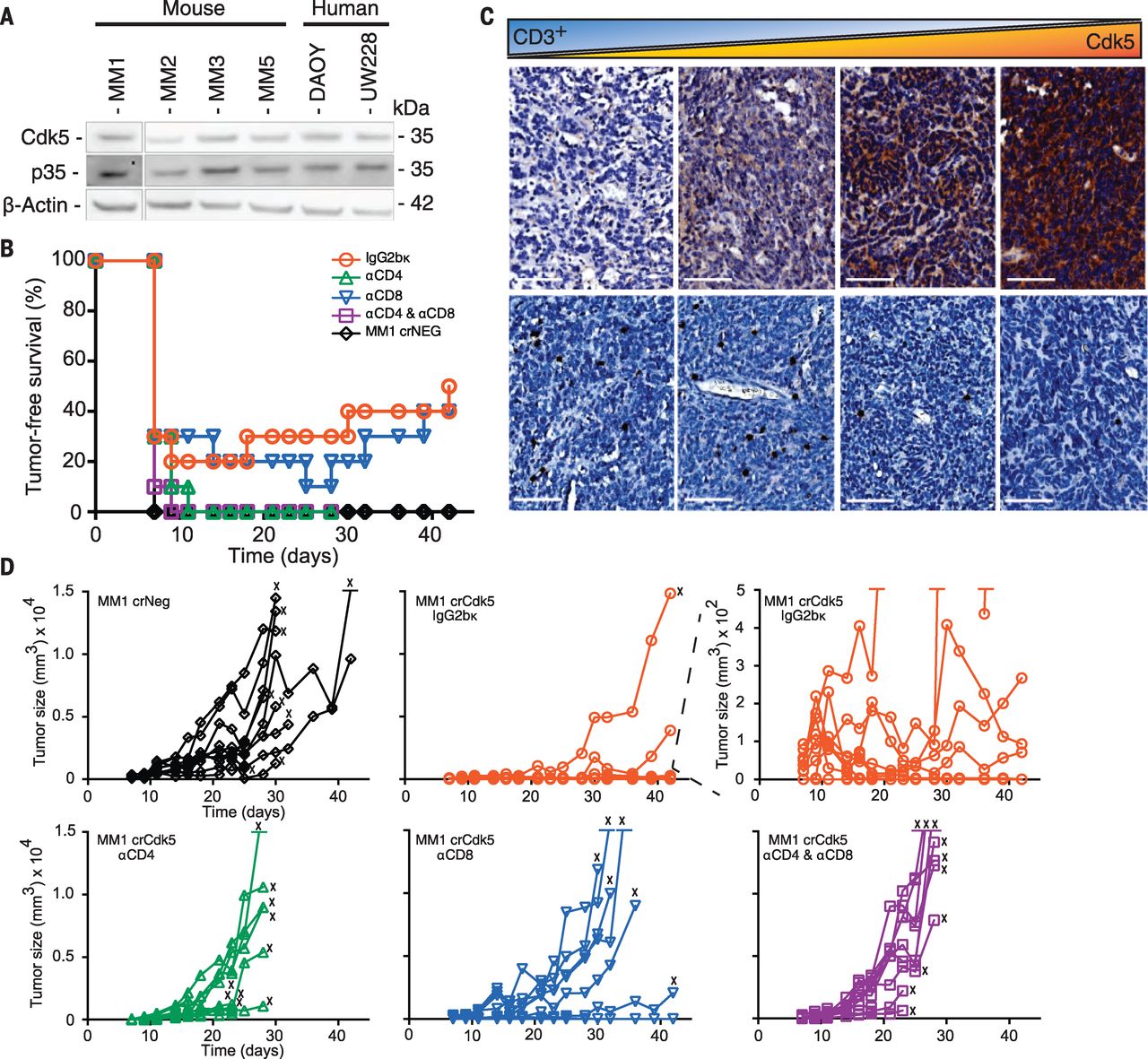 Fig. 1 Targeted deletion of Cdk5 in MB results in rejection by CD4+ T cells.[/caption]
To assess MB growth in vivo, 5 × 104 Cdk5-deficient or control cells were inoculated subcutaneously (s.c.) into the flanks of immunodeficient mice. All mice developed comparable-sized tumors by day 14. However, 78 to 50% of C57BL/6 mice injected s.c. with Cdk5-deficient MB cells showed tumor-free survival (TFS) at 19 and 42 days, whereas mice injected with WT and control tumors exhibited 0 and 7% TFS after 19 days, respectively (Fig. 1B). Mice injected with Cdk5-deficient MB cells developed significantly smaller tumors (0.02 ± 0.04 g) than mice injected with WT (0.91 ± 0.39 g) or NS (0.51 ± 0.21 g) cells. These data suggest a T cell–dependent rejection mechanism of Cdk5-deficient MM1 cells. This interpretation is supported by the observation that Cdk5 expression inversely correlated with T cell infiltration in human MB (Fig. 1C).
To identify T cell populations mediating this potent rejection, we depleted CD8+ T cells, CD4+ T cells, or both subsets in mice inoculated with MM1 crCdk5 or crNeg cells (5 × 104 s.c.). By day 11, 100% of mice injected with MM1 crNeg and 80% of mice receiving MM1 crCdk5 developed measurable tumors (Fig. 1B), although MM1 crNeg tumors were 8 times the size of MM1 crCdk5 tumors (808.8 ± 382.1 versus 101.1 ± 92.9 mm3) (Fig. 1D). Depletion with CD4-specific (αCD4) antibody alone or with both αCD4 and αCD8 antibodies resulted in 100% MM1 crCdk5 tumor incidence accompanied by rapid tumor growth, whereas CD8 depletion alone yielded 30% TFS, similar to isotype control (Fig. 1D). Among mice receiving isotype antibody, three of eight crCdk5 tumor outgrowths regressed starting on day 17, whereas three of nine crCdk5 tumor outgrowths among mice depleted of CD8+ T cells regressed starting on day 25; these outgrowths contributed to a total TFS of 50 and 40%, respectively (Fig. 1, B and D). Tumors harvested from MM1 crCdk5-bearing mice remained Cdk5– without evidence of Cdk5+ escape (fig. S4A). Similar results were seen in mice receiving MM1 shCdk5 and shNS inoculations, with a dependency on CD4+ T cells for tumor rejection. Cdk5-deficient tumors also grew aggressively in mice deficient in major histocompatibility complex class II (MHC-II). Finally, 60 to 75% of mice that rejected Cdk5-deficient tumors remained tumor free after rechallenge with a lethal dose of MM1 WT cells. Collectively, these studies point to a CD4+ T cell–dependent rejection of Cdk5-deficient tumors with robust antitumor immune memory generation.
Interferon-γ (IFN-γ) is a major CD4+ T cell effector cytokine and was abundant in Cdk5-deficient tumor mass (fig. S5A). IFN-γ induces p35, which results in enhanced Cdk5 activity. IFN-γ is known to induce PD-L1, whose expression on infiltrating immune cells is evidence of an ongoing intratumoral immune response. We examined whether disruption of Cdk5 expression in MB impaired PD-L1 induction in response to IFN-γ stimulation. We analyzed human tumor databases and found a cooccurrence of Cdk5 and PD-L1 mRNA expression in many tumor types. In Cdk5-deficient MM1, we observed a 37.58 ± 14.28% reduction in basal PD-L1 mRNA level (Fig. 2A). Note that Cdk5-deficient MM1 cells exhibited a blunted PD-L1 up-regulation in response to IFN-γ stimulation in vitro (Fig. 2, A and B). Other IFN-γ – responsive proteins, such as MHC H-2Kb and H-2Db, were not significantly affected in the Cdk5-deficient tumors, which indicated that a global disruption of the IFN-γ receptor (IFNGR) signaling was not responsible for failed PD-L1 up-regulation or enhanced immune sensitivity. Disrupting Cdk5 in rhabdomyosarcoma also led to a blunted IFN-γ−induced PD-L1 up-regulation, which indicated that the link between Cdk5 and PD-L1 regulation by IFN-γ is not MB-specific. Twenty-four hours after IFN-γ exposure, surface PD-L1 expression reached a peak of 8.2- and 6.8-fold above baseline in WT and NS cells, respectively (Fig. 2C), whereas Cdk5-deficient cells only up-regulated PD-L1 2.8-fold, so it reached a peak level similar to the basal levels in unstimulated WT and NS controls. The blunted response to IFN-γ is specific for PD-L1 but not PD-L2 (Fig. 2D). To further corroborate the link between Cdk5 and PD-L1 synthesis, we treated MM1 WT cells with roscovitine and observed a dose-dependent decrease in PD-L1 transcripts (Fig. 2E). In vitro treatment of human MB with roscovitine also diminished surface PD-L1 up-regulation with IFN-γ in a dose-dependent manner (Fig. 2F). Finally, to establish a functional link between PD-L1 and in vivo rejection of Cdk5-deficient MM1, we disrupted the PD-L1 gene (CD274) in MM1 cells (MM1 crPDL1). Similar to MM1 crCdk5 experiments, 30% of mice inoculated with MM1 crPDL1 remained tumor-free for more than 4 weeks (Figs. 2G and 1B).
[caption id="attachment_517" align="aligncenter" width="300"]
Fig. 1 Targeted deletion of Cdk5 in MB results in rejection by CD4+ T cells.[/caption]
To assess MB growth in vivo, 5 × 104 Cdk5-deficient or control cells were inoculated subcutaneously (s.c.) into the flanks of immunodeficient mice. All mice developed comparable-sized tumors by day 14. However, 78 to 50% of C57BL/6 mice injected s.c. with Cdk5-deficient MB cells showed tumor-free survival (TFS) at 19 and 42 days, whereas mice injected with WT and control tumors exhibited 0 and 7% TFS after 19 days, respectively (Fig. 1B). Mice injected with Cdk5-deficient MB cells developed significantly smaller tumors (0.02 ± 0.04 g) than mice injected with WT (0.91 ± 0.39 g) or NS (0.51 ± 0.21 g) cells. These data suggest a T cell–dependent rejection mechanism of Cdk5-deficient MM1 cells. This interpretation is supported by the observation that Cdk5 expression inversely correlated with T cell infiltration in human MB (Fig. 1C).
To identify T cell populations mediating this potent rejection, we depleted CD8+ T cells, CD4+ T cells, or both subsets in mice inoculated with MM1 crCdk5 or crNeg cells (5 × 104 s.c.). By day 11, 100% of mice injected with MM1 crNeg and 80% of mice receiving MM1 crCdk5 developed measurable tumors (Fig. 1B), although MM1 crNeg tumors were 8 times the size of MM1 crCdk5 tumors (808.8 ± 382.1 versus 101.1 ± 92.9 mm3) (Fig. 1D). Depletion with CD4-specific (αCD4) antibody alone or with both αCD4 and αCD8 antibodies resulted in 100% MM1 crCdk5 tumor incidence accompanied by rapid tumor growth, whereas CD8 depletion alone yielded 30% TFS, similar to isotype control (Fig. 1D). Among mice receiving isotype antibody, three of eight crCdk5 tumor outgrowths regressed starting on day 17, whereas three of nine crCdk5 tumor outgrowths among mice depleted of CD8+ T cells regressed starting on day 25; these outgrowths contributed to a total TFS of 50 and 40%, respectively (Fig. 1, B and D). Tumors harvested from MM1 crCdk5-bearing mice remained Cdk5– without evidence of Cdk5+ escape (fig. S4A). Similar results were seen in mice receiving MM1 shCdk5 and shNS inoculations, with a dependency on CD4+ T cells for tumor rejection. Cdk5-deficient tumors also grew aggressively in mice deficient in major histocompatibility complex class II (MHC-II). Finally, 60 to 75% of mice that rejected Cdk5-deficient tumors remained tumor free after rechallenge with a lethal dose of MM1 WT cells. Collectively, these studies point to a CD4+ T cell–dependent rejection of Cdk5-deficient tumors with robust antitumor immune memory generation.
Interferon-γ (IFN-γ) is a major CD4+ T cell effector cytokine and was abundant in Cdk5-deficient tumor mass (fig. S5A). IFN-γ induces p35, which results in enhanced Cdk5 activity. IFN-γ is known to induce PD-L1, whose expression on infiltrating immune cells is evidence of an ongoing intratumoral immune response. We examined whether disruption of Cdk5 expression in MB impaired PD-L1 induction in response to IFN-γ stimulation. We analyzed human tumor databases and found a cooccurrence of Cdk5 and PD-L1 mRNA expression in many tumor types. In Cdk5-deficient MM1, we observed a 37.58 ± 14.28% reduction in basal PD-L1 mRNA level (Fig. 2A). Note that Cdk5-deficient MM1 cells exhibited a blunted PD-L1 up-regulation in response to IFN-γ stimulation in vitro (Fig. 2, A and B). Other IFN-γ – responsive proteins, such as MHC H-2Kb and H-2Db, were not significantly affected in the Cdk5-deficient tumors, which indicated that a global disruption of the IFN-γ receptor (IFNGR) signaling was not responsible for failed PD-L1 up-regulation or enhanced immune sensitivity. Disrupting Cdk5 in rhabdomyosarcoma also led to a blunted IFN-γ−induced PD-L1 up-regulation, which indicated that the link between Cdk5 and PD-L1 regulation by IFN-γ is not MB-specific. Twenty-four hours after IFN-γ exposure, surface PD-L1 expression reached a peak of 8.2- and 6.8-fold above baseline in WT and NS cells, respectively (Fig. 2C), whereas Cdk5-deficient cells only up-regulated PD-L1 2.8-fold, so it reached a peak level similar to the basal levels in unstimulated WT and NS controls. The blunted response to IFN-γ is specific for PD-L1 but not PD-L2 (Fig. 2D). To further corroborate the link between Cdk5 and PD-L1 synthesis, we treated MM1 WT cells with roscovitine and observed a dose-dependent decrease in PD-L1 transcripts (Fig. 2E). In vitro treatment of human MB with roscovitine also diminished surface PD-L1 up-regulation with IFN-γ in a dose-dependent manner (Fig. 2F). Finally, to establish a functional link between PD-L1 and in vivo rejection of Cdk5-deficient MM1, we disrupted the PD-L1 gene (CD274) in MM1 cells (MM1 crPDL1). Similar to MM1 crCdk5 experiments, 30% of mice inoculated with MM1 crPDL1 remained tumor-free for more than 4 weeks (Figs. 2G and 1B).
[caption id="attachment_517" align="aligncenter" width="300"]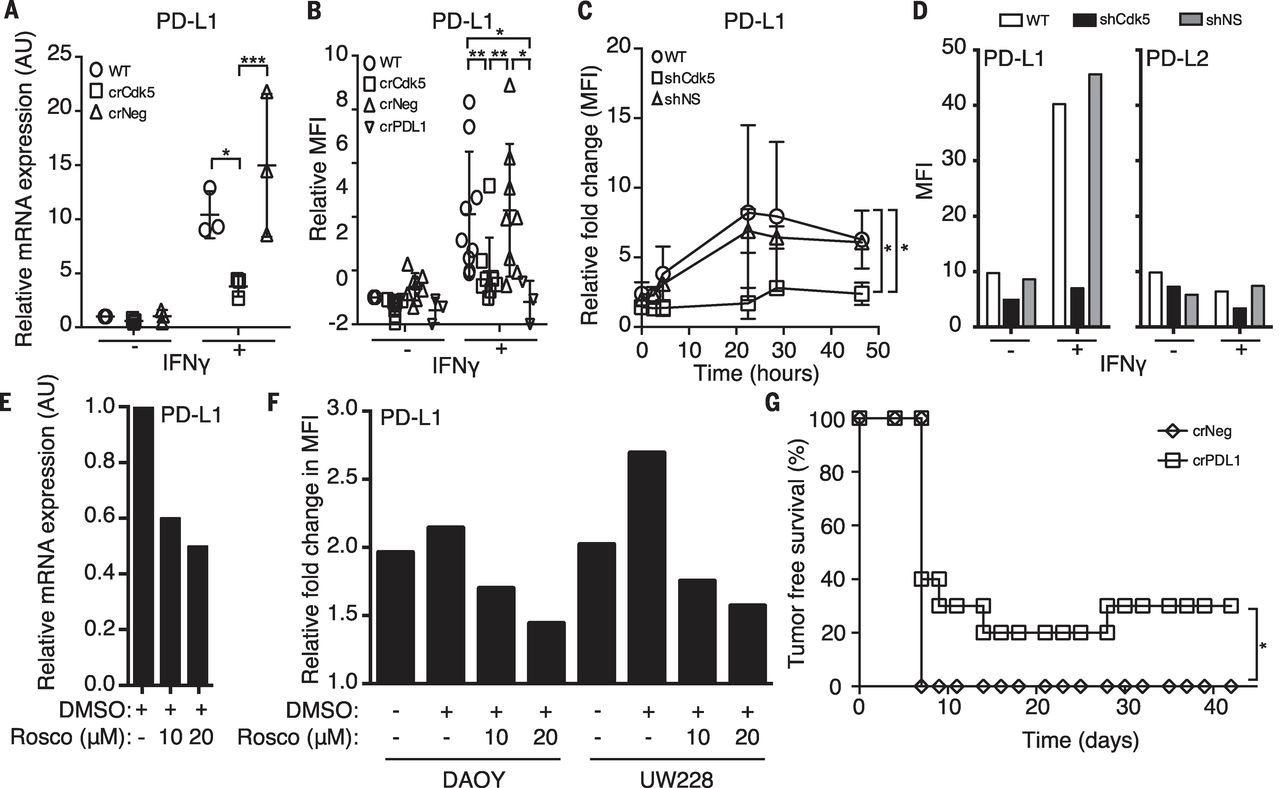 Fig. 2 Disruption of either Cdk5 gene expression or Cdk5 activity suppresses PD-L1 expression that cannot be overcome with IFN-γ stimulation in both human and murine MBs.[/caption]
Next, we interrogated the IFNGR signaling pathway. Western blot analysis of various MM1 cells failed to show differences, after IFN-γ exposure, in STAT1, STAT2, or STAT3 (members of the family of signal transducers and activators of transcription) (Fig. 3A), in agreement with the robust MHC class I induction in Cdk5-deficient MM1 cells (fig. S7A). To further dissect this STAT1-independent signaling, we examined interferon regulatory factor-1 (IRF1) and interferon regulatory factor-2 (IRF2), which are implicated as positive and negative regulators of PD-L1 transcription, respectively. IRF1 protein was rapidly induced by IFN-γ and remained elevated for up to 48 hours regardless of Cdk5 expression (Fig. 3A). We observed a rapid loss of the PD-L1 transcription repressor, IRF2, in WT and crNeg cells. In contrast, IRF2 and its corepressor IRF2BP2 were elevated at baseline in Cdk5-deficient cells and persisted for up to 48 hours after IFN-γ exposure (Fig. 3A). This protein expression difference cannot be accounted for at the transcriptional level. Phosphoproteomic analysis identified 77 distinct phosphopeptides in the shCdk5 versus WT or shNS screen, and 798 phosphopeptides in the crCdk5 versus WT or crNeg screen. Between these two data sets, 22 common proteins were differentially phosphorylated in Cdk5-deficient cells, with IRF2BP2 among the highest phosphorylated peptide species (Fig. 3B).
[caption id="attachment_521" align="aligncenter" width="300"]
Fig. 2 Disruption of either Cdk5 gene expression or Cdk5 activity suppresses PD-L1 expression that cannot be overcome with IFN-γ stimulation in both human and murine MBs.[/caption]
Next, we interrogated the IFNGR signaling pathway. Western blot analysis of various MM1 cells failed to show differences, after IFN-γ exposure, in STAT1, STAT2, or STAT3 (members of the family of signal transducers and activators of transcription) (Fig. 3A), in agreement with the robust MHC class I induction in Cdk5-deficient MM1 cells (fig. S7A). To further dissect this STAT1-independent signaling, we examined interferon regulatory factor-1 (IRF1) and interferon regulatory factor-2 (IRF2), which are implicated as positive and negative regulators of PD-L1 transcription, respectively. IRF1 protein was rapidly induced by IFN-γ and remained elevated for up to 48 hours regardless of Cdk5 expression (Fig. 3A). We observed a rapid loss of the PD-L1 transcription repressor, IRF2, in WT and crNeg cells. In contrast, IRF2 and its corepressor IRF2BP2 were elevated at baseline in Cdk5-deficient cells and persisted for up to 48 hours after IFN-γ exposure (Fig. 3A). This protein expression difference cannot be accounted for at the transcriptional level. Phosphoproteomic analysis identified 77 distinct phosphopeptides in the shCdk5 versus WT or shNS screen, and 798 phosphopeptides in the crCdk5 versus WT or crNeg screen. Between these two data sets, 22 common proteins were differentially phosphorylated in Cdk5-deficient cells, with IRF2BP2 among the highest phosphorylated peptide species (Fig. 3B).
[caption id="attachment_521" align="aligncenter" width="300"]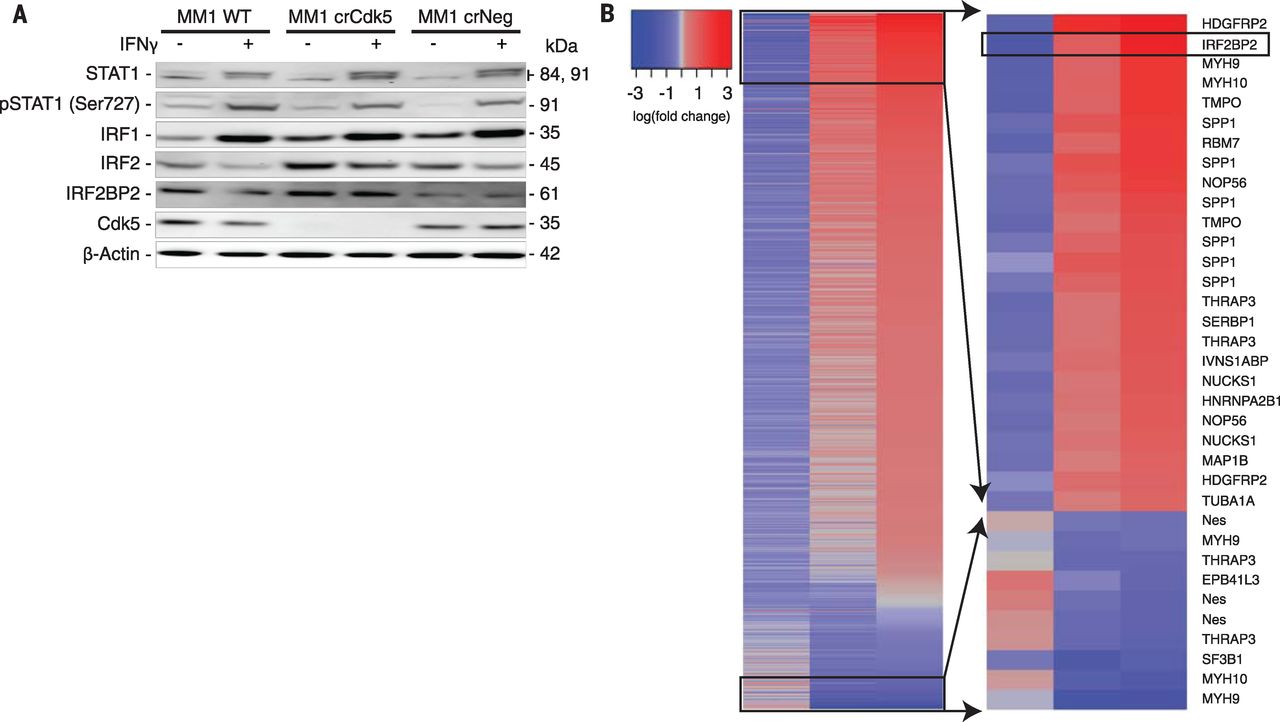 Fig. 3 Cdk5 gene silencing alters the IFN-γ signaling pathway and is associated with hyperphosphorylation of IRF2BP2.[/caption]
Finally, we introduced Cdk5-deficient MM1 cells orthotopically into C57BL/6 mice. Gross inspection revealed a 50% tumor incidence in mice injected with Cdk5-deficient MM1, mirroring s.c. tumors. In contrast, 100% of mice injected with WT or NS MM1 cells developed gross brain tumors by day 14. Intracranial (i.c.) Cdk5-deficient tumor outgrowth remained devoid of Cdk5 expression without the emergence of a Cdk5+ escape variant. Histological analysis showed increased accumulation of IBA-1+ cells, which marks microglia and infiltrating monocytes, and PD-L1+ staining in the Cdk5-deficient MM1 tumor margin and surrounding stroma (Fig. 4A). Immune cell composition analysis showed a modest increase in CD3+ T cells, similar to that shown by immunohistochemical data (Fig. 1C). However, the percentages of CD3+ cells were equivalent in crCdk5 and WT tumor samples by flow cytometry (Fig. 4B). Cdk5-deficient tumors elicited an increased ratio of CD8+ to CD4+ T cell infiltrate, lower PD-1 expression in CD4+ T cells, and higher PD-L1 expression in both T cell subsets (Fig. 4, C to E). Although CD8+ T cells are not the primary antitumor effector cells in this model, their increased recruitment likely reflects an overall inflammatory tumor milieu as evidenced by increased PD-L1 expression and overall tissue IFN-γ levels (Fig. 1B). The myeloid infiltrate in i.c. tumors shifted from a Ly6C– to a Ly6Chi population with an increased percentage of PD-L1+ cells in bulk CD11b+ cells and in each Ly6C subset (Fig. 4, F to H), accompanied by a decrease in the percentage of microglia (CD11b+CD45lo) (Fig. 4F). The Ly6Clo subset expressed a higher density of surface PD-L1 in the Cdk5-deficient tumors (Fig. 4H). Again, this finding was recapitulated in s.c. tumors, which showed a significant increase in the percentage of PD-L1+immune cells, with a trend toward increased density of PD-L1 staining in the crCdk5 tumor microenvironment. The observed increase in PD-L1+ populations and staining density aligns with histologic analyses (Fig. 4A), which suggests a state of global immune activation in response to ongoing IFN-γ stimulation. This finding is in good agreement with reports showing increased PD-L1+ immune cells in MB stroma undergoing active immune checkpoint blockade.
[caption id="attachment_522" align="aligncenter" width="300"]
Fig. 3 Cdk5 gene silencing alters the IFN-γ signaling pathway and is associated with hyperphosphorylation of IRF2BP2.[/caption]
Finally, we introduced Cdk5-deficient MM1 cells orthotopically into C57BL/6 mice. Gross inspection revealed a 50% tumor incidence in mice injected with Cdk5-deficient MM1, mirroring s.c. tumors. In contrast, 100% of mice injected with WT or NS MM1 cells developed gross brain tumors by day 14. Intracranial (i.c.) Cdk5-deficient tumor outgrowth remained devoid of Cdk5 expression without the emergence of a Cdk5+ escape variant. Histological analysis showed increased accumulation of IBA-1+ cells, which marks microglia and infiltrating monocytes, and PD-L1+ staining in the Cdk5-deficient MM1 tumor margin and surrounding stroma (Fig. 4A). Immune cell composition analysis showed a modest increase in CD3+ T cells, similar to that shown by immunohistochemical data (Fig. 1C). However, the percentages of CD3+ cells were equivalent in crCdk5 and WT tumor samples by flow cytometry (Fig. 4B). Cdk5-deficient tumors elicited an increased ratio of CD8+ to CD4+ T cell infiltrate, lower PD-1 expression in CD4+ T cells, and higher PD-L1 expression in both T cell subsets (Fig. 4, C to E). Although CD8+ T cells are not the primary antitumor effector cells in this model, their increased recruitment likely reflects an overall inflammatory tumor milieu as evidenced by increased PD-L1 expression and overall tissue IFN-γ levels (Fig. 1B). The myeloid infiltrate in i.c. tumors shifted from a Ly6C– to a Ly6Chi population with an increased percentage of PD-L1+ cells in bulk CD11b+ cells and in each Ly6C subset (Fig. 4, F to H), accompanied by a decrease in the percentage of microglia (CD11b+CD45lo) (Fig. 4F). The Ly6Clo subset expressed a higher density of surface PD-L1 in the Cdk5-deficient tumors (Fig. 4H). Again, this finding was recapitulated in s.c. tumors, which showed a significant increase in the percentage of PD-L1+immune cells, with a trend toward increased density of PD-L1 staining in the crCdk5 tumor microenvironment. The observed increase in PD-L1+ populations and staining density aligns with histologic analyses (Fig. 4A), which suggests a state of global immune activation in response to ongoing IFN-γ stimulation. This finding is in good agreement with reports showing increased PD-L1+ immune cells in MB stroma undergoing active immune checkpoint blockade.
[caption id="attachment_522" align="aligncenter" width="300"]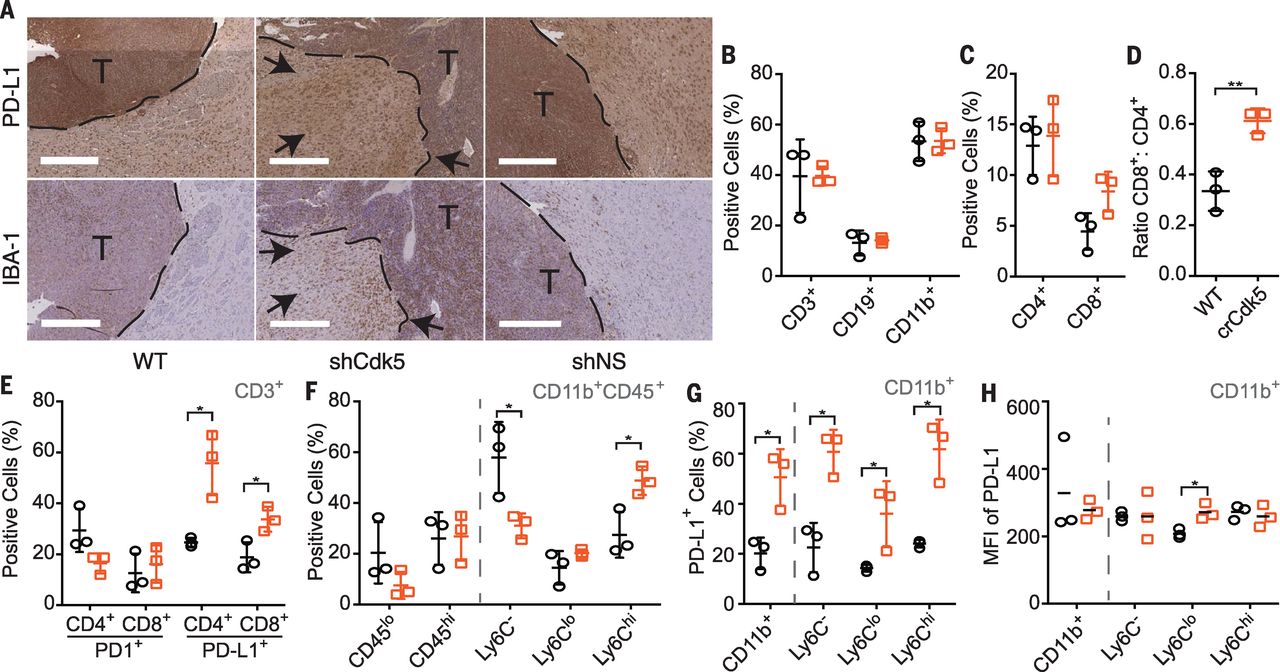 Fig. 4 Orthotopic Cdk5-deficient tumors exhibit increased PD-L1 staining, CD4+tumor–infiltrating lymphocytes (TILs), and accumulating infiltrates of CD11b+ populations.[/caption]
Here, we showed that Cdk5 disruption sensitizes MB to CD4+ T cell–dependent rejection via posttranslational modification of IRF2BP2, which increases IRF2 and IRF2BP2 abundance and sustains PD-L1 transcriptional repression after IFN-γ stimulation. Downstream IFN-γ signaling induces interferon-stimulated genes, including IRF1, which activates secondary-response genes, including PD-L1. IRF2 acts as a repressor that competes with IRF1 for binding to the same promoter element. Constitutively present, IRF2 is up-regulated in response to either type I IFNs or IRF1 and provides a negative-feedback loop by binding to its own promoter to block transcription. The prolonged half-life of IRF2 (8 hours) relative to IRF1 (0.5 hours) provides a mechanism for IRF2 antagonism. IRF2BP2 was recently identified as a corepressor with IRF2, and low IRF2BP2 expression was correlated with high PD-L1 expression in breast cancer. Our data provide a direct link between disruption of Cdk5 activity and IRF2BP2 hyperphosphorylation at sites that are distinct from previously described sites that affect nuclear localization, vascular endothelial growth factor A, or MHC-I expression, which suggests that Cdk5 either directly or indirectly inhibits other kinase(s) that phosphorylate IRF2BP2.
PD-L1 and PD-1 play a critical role in tumor immune evasion, with ~30% of tumors responding to immune checkpoint blockade. High Cdk5 expression correlates with worse clinical outcome in multiple cancers. In our studies, both Cdk5- and PD-L1-deficient MB cells exhibit similar TFS (Figs. 1B and 2G). More CD4+ T cells with lower PD-1 expression were found in the Cdk5-deficient CNS tumors, whereas CD11b+ cells accumulate in larger quantities with higher PD-L1+ expression (Fig. 4, C to G). Myeloid PD-L1 up-regulation may be a response to overall increased IFN-γ. Alternatively, these cells may play a distinct role modulating infiltrating T cell function, which are present in most human MB specimens. Last, as Cdk5 directly phosphorylates MYC on Ser62 , it remains to be determined whether Cdk5 plays a role in MYC-regulated PD-L1 expression.
Fig. 4 Orthotopic Cdk5-deficient tumors exhibit increased PD-L1 staining, CD4+tumor–infiltrating lymphocytes (TILs), and accumulating infiltrates of CD11b+ populations.[/caption]
Here, we showed that Cdk5 disruption sensitizes MB to CD4+ T cell–dependent rejection via posttranslational modification of IRF2BP2, which increases IRF2 and IRF2BP2 abundance and sustains PD-L1 transcriptional repression after IFN-γ stimulation. Downstream IFN-γ signaling induces interferon-stimulated genes, including IRF1, which activates secondary-response genes, including PD-L1. IRF2 acts as a repressor that competes with IRF1 for binding to the same promoter element. Constitutively present, IRF2 is up-regulated in response to either type I IFNs or IRF1 and provides a negative-feedback loop by binding to its own promoter to block transcription. The prolonged half-life of IRF2 (8 hours) relative to IRF1 (0.5 hours) provides a mechanism for IRF2 antagonism. IRF2BP2 was recently identified as a corepressor with IRF2, and low IRF2BP2 expression was correlated with high PD-L1 expression in breast cancer. Our data provide a direct link between disruption of Cdk5 activity and IRF2BP2 hyperphosphorylation at sites that are distinct from previously described sites that affect nuclear localization, vascular endothelial growth factor A, or MHC-I expression, which suggests that Cdk5 either directly or indirectly inhibits other kinase(s) that phosphorylate IRF2BP2.
PD-L1 and PD-1 play a critical role in tumor immune evasion, with ~30% of tumors responding to immune checkpoint blockade. High Cdk5 expression correlates with worse clinical outcome in multiple cancers. In our studies, both Cdk5- and PD-L1-deficient MB cells exhibit similar TFS (Figs. 1B and 2G). More CD4+ T cells with lower PD-1 expression were found in the Cdk5-deficient CNS tumors, whereas CD11b+ cells accumulate in larger quantities with higher PD-L1+ expression (Fig. 4, C to G). Myeloid PD-L1 up-regulation may be a response to overall increased IFN-γ. Alternatively, these cells may play a distinct role modulating infiltrating T cell function, which are present in most human MB specimens. Last, as Cdk5 directly phosphorylates MYC on Ser62 , it remains to be determined whether Cdk5 plays a role in MYC-regulated PD-L1 expression.