Ellefson et al. construct a reverse transcriptase using in vitro directed evolution and protein engineering from a prokaryotic DNA polymerase. This reverse transcriptase shows increased fidelity as compared to natural reverse transcriptases and increases the precision of transcriptomics methods. Ellefson et al. published their results in Science Magazine (Science 24 Jun 2016: Vol. 352, Issue 6293, pp. 1590-1593 DOI: 10.1126/science.aaf5409).
The article is followed:
The molecular basis for life rests on the information flow between DNA, RNA, and proteins. Early notions of a unidirectional central dogma were amended after the discovery of the reverse transcriptase (RT) enzyme. The RT family has a single ancient evolutionary origin based on amino acid homology and the presence of RT across multiple domains of life. RTs are involved in processes such as telomere addition, mitochondrial plasmid replication, transposition, and the proliferation of retroviral genomes. It is also hypothesized to be the catalyst in the transition of the RNA to DNA world by providing an avenue to copy RNA into more stable DNA genomes.
The progenitor of RT is postulated to be an RNA-dependent RNA polymerase. Because RNA polymerases generally lack an error-checking 3′-5′ exonuclease domain, proofreading activity is also not present across the RT family, resulting in low-fidelity reverse transcription and characteristic quasispecies behavior in organisms that rely upon it for replication. In contrast to RTs, other DNA polymerase families have evolved exquisite proofreading mechanisms to increase DNA synthesis fidelity during genome replication.
To determine whether the evolutionary divide between RTs and DNA polymerases is a matter of history or function, we have attempted to directly evolve a reverse transcription xenopolymerase (RTX; Fig. 1A) from an error-correcting DNA polymerase using a modified directed evolution strategy, reverse transcription–compartmentalized self-replication (RT-CSR) (Fig. 1B). RT-CSR enables the simultaneous screening of up to 109 polymerase variants for RT activity.
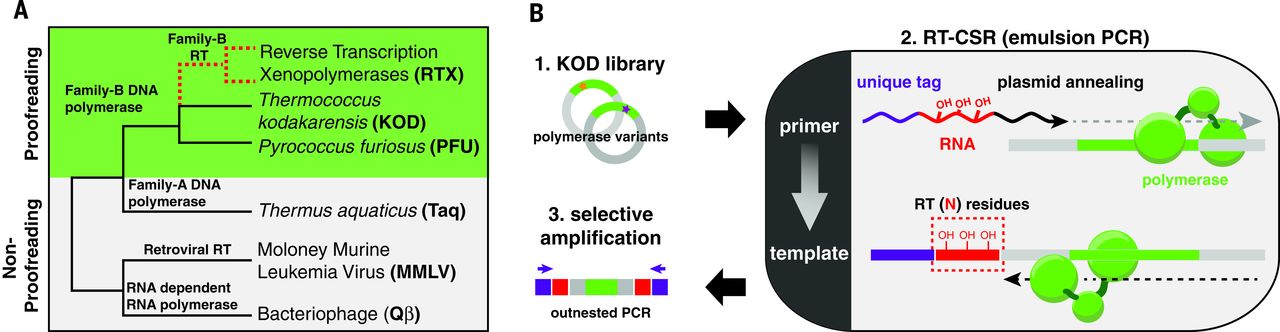
Fig. 1 Evolution of a synthetic family of reverse transcriptases by RT-CSR.
We chose the Archaeal family-B DNA polymerases (polB) for directed evolution of the RTX as they are monomeric, hyperthermostable, highly processive, and contain proofreading domains. Attempts to rationally design these enzymes to use RNA templates have met with limited success, and initial experiments confirmed that two common polB enzymes from Pyrococcus furiosus and Thermococcus kodakarensis (KOD) failed to polymerize across five template RNA bases. Modeling to identify mutations enabling RT activity was deemed impractical, given the extensive contacts these polymerases make with the template (>50 direct interactions). We initiated evolution using low-stringency RT-CSR (10 RNA residues) with a random library (one or two amino acid mutations per gene) of KOD polymerase variants. As polymerases were enriched, we gradually increased RT-CSR stringency with the stepwise addition of RNA bases into primers. By cycle 18, primers were entirely composed of RNA—requiring reverse transcription of 176 residues to occur every thermal cycle to maintain exponential amplification in the emulsion polymerase chain reaction (PCR).
Profiling of polymerases revealed one variant, B11, which contained 37 mutations. RT-CSR enriched for RT activity, and B11 was capable of reverse transcription of at least 500 base pairs; however, sequencing and testing confirmed inactivation of the proofreading domain. Kinetic analyses established that B11 uses both DNA and RNA templates with similar efficiencies by greatly lowering the Michaelis constant (Km) on RNA: DNA heteroduplexes. We attempted to restore proofreading by transplantation of the wild-type 3′-5′ exonuclease, which reactivated proofreading capabilities, albeit to barely detectable levels. Encouraged that minimizing extraneous mutations could restore proofreading activity, we sought to design polymerases with a minimal set of mutations.
To understand how our process reshaped KOD polymerase to use RNA templates, we deep-sequenced RT-CSR cycles to recapitulate the evolutionary path to RT activity (Fig. 2A). Mutations were identified throughout the polymerase and accumulated along the template-binding interface so as to progressively increase the length of RNA that could be accommodated. The mutated positions are hypothesized to be molecular checkpoints used to enforce strict DNA template utilization: as the template enters, near the active site, and at the nascent duplex. Given the likely importance of these regions, we used computer modeling to determine the molecular basis for RNA utilization.
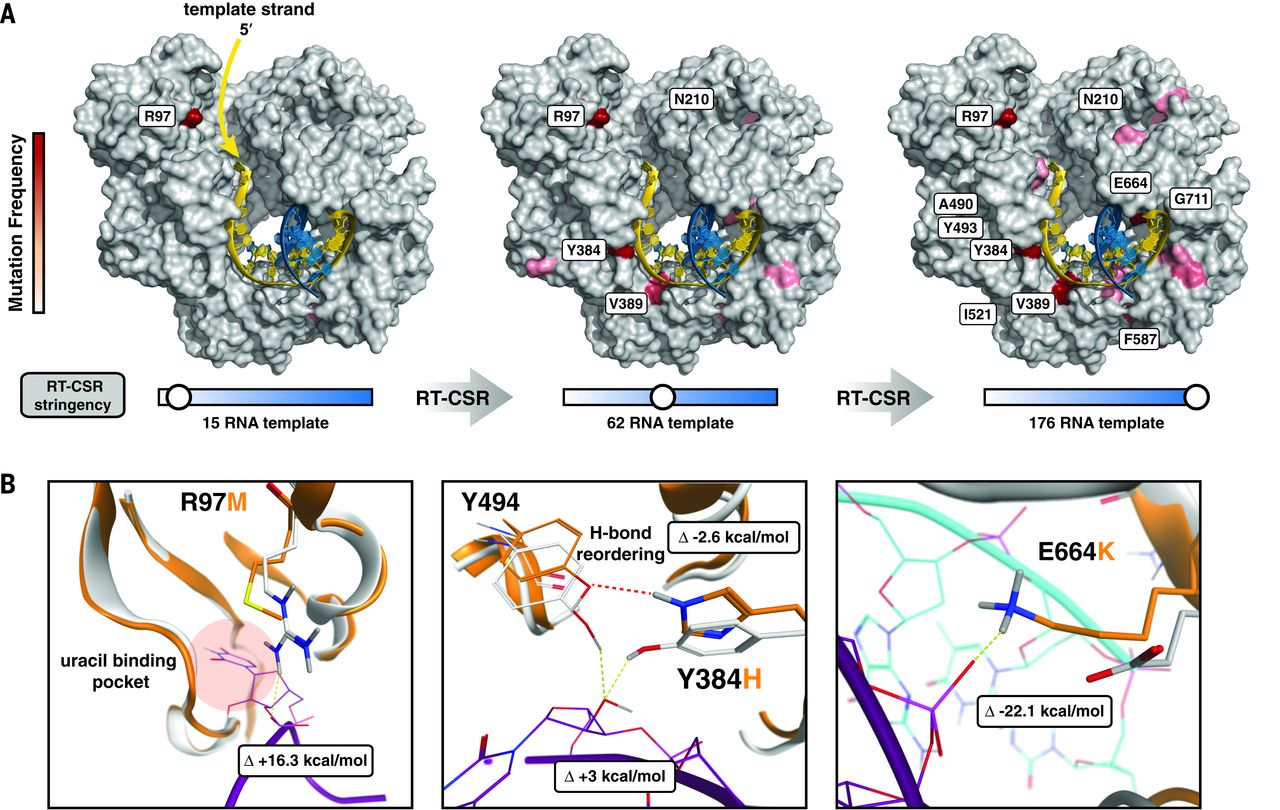
Fig. 2 Molecular checkpoints involved in template recognition.
The first selected mutation localized near the template entry site of the polymerase at position R97 (Fig. 2B). Proximal to this site, native polB scans for uracils (typically caused by cytosine deamination) by flipping template bases into a specialized pocket to halt polymerization until the mutation can be corrected by repair machinery. Evolved polymerases contained a variety of amino acid mutations at R97, all of which destabilize a salt bridge to the phosphate backbone that presumably regulates base flipping into the pocket.
As template residues near the active site, they encounter mutation Y384H, which prevents Y384 and Y494 from hydrogen bonding to the 2′ hydroxyl of template RNA by reorganizing a hydrogen bonding network. After polymerization, in the thumb domain, the most prevalent mutations (E664K, G711V, and E735K) promote tighter homo- and heteroduplex binding in both A- and B-form conformations. The E664K mutation alone has been shown to increase binding to RNA:DNA heteroduplexes. To further validate that we had established an optimized set of mutations, we fully randomized several positions and repeated the RT-CSR. In support of our modeling, many amino acids solutions were viable at position R97, but other positions (Y384 and E664) had strong preferences for particular amino acids.
Modeled designs of several polymerases with favorable RT mutations were synthesized and tested empirically. The best-performing RTX contained fewer than half the mutations found in B11, without sacrificing catalytic efficiency or Km on RNA. Mutations in RTX did not affect desirable properties of parental KOD polymerase. Thermostability was maintained, with optimal RT occurring at ~70 °C, and consequently RTX was capable of single-enzyme RT-PCR (in which RTX performs both first-strand RT synthesis and PCR amplification). Across several RNA samples and gene loci, RTX demonstrated high processivity on RNA templates, performing RT-PCR on RNAs more than 5 kb in length.
Initial testing of RTX using dideoxy mismatch primers in PCR demonstrated robust proofreading activity on DNA template, but it was unclear whether the proofreading mechanism was compatible during reverse transcription because RNA:DNA heteroduplexes can adopt A-form helical structures. Primer extension reactions with a canonical matched base pair or a 3′ deoxy mismatched pair (preventing extension until terminator excision) were tested. Both wild-type KOD and RTX were capable of extending mismatched primers on DNA templates, unlike exonuclease-deficient mutants. When tested on an RNA template, KOD’s exonuclease was stimulated—actively degrading the priming oligonucleotide. In contrast, RTX could extend the mismatched primer with activity indistinguishable from that of DNA templated proofreading (Fig. 3A).
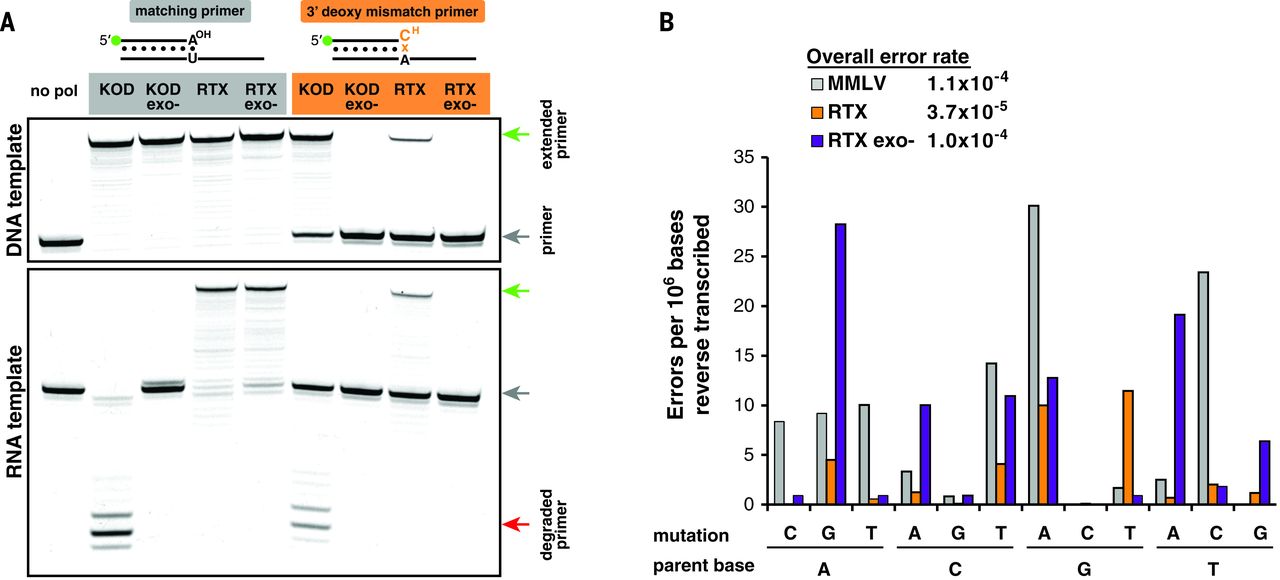
Fig. 3 RTX polymerase proofreads during reverse transcription.
Given that RTX is capable of proofreading during reverse transcription, we hypothesized that it may have increased RT fidelity compared to natural polymerases. Barcoded primers used during RT of several human mRNAs allowed multiple reads of a single cDNA during deep sequencing—reducing background sequencing errors by several orders of magnitude. Sequencing analyses revealed that the control retroviral RT [Moloney murine leukemia virus (MMLV)] had an error rate of 1.1 × 10−4 to 4.8 × 10−4, whereas RTX had an error rate of 3.5 × 10−5 to 3.7 × 10−5 (3- to 10-fold lower). The mutational spectra of RTX favored G-to-A transitions and G-to-T transversions, which accounted for nearly half the observed mutations. Inactivating the RTX’s proofreading capabilities increased error frequency nearly threefold, supporting evidence that active proofreading was occurring during RT. Inactivating the proofreading of RTX shifted the mutational bias (Fig. 3B). Given that the barcoding error detection limit is identical to the observed error of RTX, we anticipate the true error rate for RTX to be even lower than reported.
RTX has the potential to streamline workflows (combining RT and PCR steps) and increase the precision of transcriptomics, reducing biases and errors introduced in the reverse transcription step of RNA-sequencing protocols. To demonstrate its utility, we introduced RTX into a commonly used platform for RNA sequencing. Analysis revealed nearly identical coverage and expression profiles, suggesting that RTX is compatible with established workflows. In addition, we developed a more streamlined protocol to directly sequence RNA. Using a traditional Sanger sequencing approach, we directly sequenced a GATC5 RNA repeat. Direct RNA sequencing should be adaptable to single-molecule sequencing platforms, enabling high-throughput and high-fidelity sequencing of complex RNA samples by eliminating the biases created in cDNA synthesis and subsequent amplification.
The expanded template specificity of the RTX lineage may presage the ability to use entirely new chemistries in genetics. Primer extension reactions were performed on a ribose sugar analog [2′ O-methyl (Me) DNA] that indicated that RTX reverse transcription could extend alternative templates but with much lower efficiency, indicating a preference for RNA substrates. However, RTX was still far more efficient at using 2′-OMe DNA than the parental wild-type, allowing the possibility of further optimization and, owing to 2′-OMe stability, potential therapeutic applications.
The RTX RNA reverse transcriptase function is fundamentally distinct from that of the retroelement lineage. Using RT-CSR, we have altered the substrate specificity of a high-fidelity DNA polymerase, highlighting the plasticity of highly conserved molecular machinery. Ostensibly, the mutations identified unlocked molecular checkpoints in the discrimination of DNA and RNA, but did not disrupt the proofreading capabilities of the polymerase. This was unexpected, especially given that RNA:DNA hybrid duplexes often form A-helical structures unlike DNA:DNA duplexes, and may provide insights into the transition from polymerization to editing modes of the polymerase.
Only a handful of mutations were required to impart RT activity, suggesting that the evolutionary hurdle for forming high-fidelity reverse transcription is relatively low. Nevertheless, all known retroelements use proofreading-deficient RTs, suggesting that high error rates are either a historical coincidence or an evolutionary strategy to promote diversity. Another possible explanation is that high fidelity was never required simply because RNA genomes are small as a result of their inherent instability. Given the plasticity of these polymerases for modified templates and the adaptability of the RT-CSR framework (as primers are simply programmed to contain modified bases), RTX evolution should be compatible with many base and sugar analogs. Combination with previously evolved XNA polymerases could enable synthesis of genomes entirely composed of artificial nucleic acids.
Reference:
http://science.sciencemag.org/content/352/6293/1590
[table caption=”Related Products” width=”800″ colwidth=”100|250|150″ colalign=”left|left|center|center|center”]
Cat. #,Product name,Source(Host),Species,Conjugate
CMC03,M-MLV H- Reverse Transcriptase,/,/,/
TERT-16646M,Recombinant Mouse TERT Protein,Mammalian Cells,Mouse,His
TERT-374H,Recombinant Human TERT,Mammalian Cells,Human,His
TERT-2688C,Recombinant Chicken TERT,Mammalian Cells,Chicken,His
TERT-5473Z,Recombinant Zebrafish TERT,Mammalian Cells,Zebrafish,His
[/table]