Pancreatic ductal adenocarcinoma (PDAC) is the most common malignancy of the pancreas. PDAC is an aggressive and difficult malignancy to treat. Incidence of PDAC correlates with increasing age and therefore is an increasing problem as world population is aging. By 2030, researchers project that pancreatic cancer will become the 2nd leading cause of cancer related death in the US after lung cancer, surpassing colorectal, breast, and prostate cancer .
Chromosomal region 18q21, which contains SMAD4, is homozygously deleted in a large fraction of human solid tumours, including PDAC.
A recent study analyzed the copy-number, tanscriptomic and microdissected PDAC samples, and showed a stron positive correlation between ME2 and SMAD4, which indicated that ME2 is often deleted along with SMAD4, which is the housekeeping gene contained in the 18q21 region. Furthermore, immunohistochemistry (IHC) analysis of ME2 and SMAD4 expression in 62 PDAC and 77 matched normal samples revealed corresponding loss of both proteins in 37% of PDAC cases. Beyond PDAC, homozygous deletion of ME2 occurs in many other cancers.
Malate dehydrogenase (decarboxylating) or NAD-malic enzyme (NAD-ME) is an enzyme that catalyzes the chemical reaction:
(S)-malate + NAD+ <=> pyruvate + CO2 + NADH
NADP-dependent malic enzyme is a protein that in humans is encoded by the ME1 gene. This gene encodes a cytosolic, NADP-dependent enzyme that generates NADPH for fatty acid biosynthesis. The activity of this enzyme, the reversible oxidative decarboxylation of malate to pyruvate, links the glycolytic and citric acid cycles. Mammalian tissues contain 3 distinct isoforms of malic enzyme: a cytosolic NADP(+)-dependent isoform (ME1), a mitochondrial NADP(+)-dependent isoform (ME3), and a mitochondrial NAD(+)-dependent isoform (ME2).
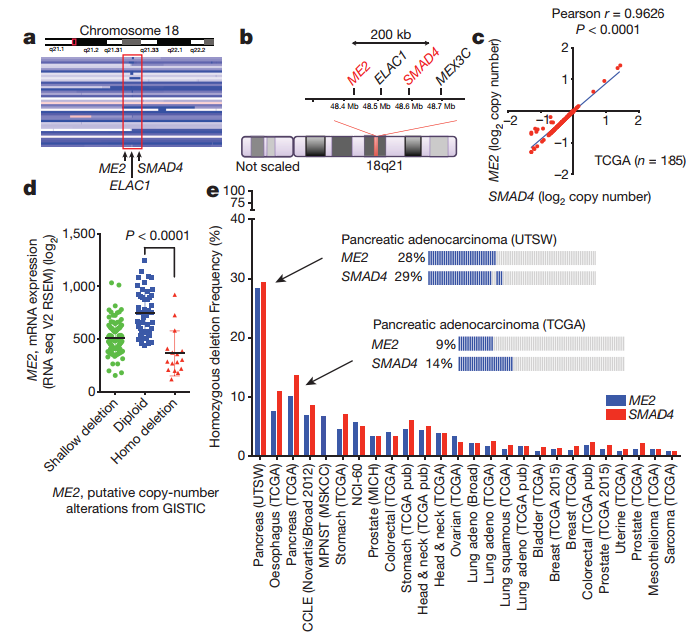
Fig1. ME2 is often deleted along with SMAD4 in PDAC
In normal cells and most cancer cells, ME2 is the main isoform in mitochondria and has higher abundance and enzymatic activity compared to ME3. However, in the situation of absence of ME2, ME3 presumably sustains essential NADPH synthesis in mitochondria.
In that study, the author hypothesis that ME3 could support ME2-deficient cell survive, and analysis the ME2 and ME3 expression level in ME2 homozygous null and ME2-intact cell lines and in matched normal and PDAC samples. The assay results suggest that ME1 might not be a viable collateral lethality target; mitochondrial malic enzymes have redundant and cell-essential roles and the depletion of ME3 in ME2-null PDAC cells could lead to collateral lethality.
Dox-induction of ME3 shRNA in ME2-null PDAC tumors decreased colony formation, proliferation and increased cell apoptosis, which phenotypes could alleviate by ectopic expression of ME2 in mitochondrial ME3-depleted cells. Cell-permeable reduced glutathione (GSH) or antioxidant N-acetylcysteine (NAC) could partially rescue cell proliferation for ME3 depleted ME2-null cell and cell-permeable pyruvate could partially rescue cell proliferation and decrease ROS both.
ME3 depletion in ME2-null cells led to an increase in Gln flux via the TCA cycle, Glc flux to lactate and pyruvate was unchanged following, but Glc entry into the TCA cycle was decreased upon depletion of mitochondrial malic enzymes. Besides, ME3-depleted also decreased oxygen consumption rate (OCR). The Mito Tracker staining and TEM showed that mitochondrial structure changed under ME3 depletion, which has more globular appearances and aberrant ring-shape, which is a feature consistent with ROS mediated stress. These data suggest that ME3 depletion leads to inhibition of mitochondrial function.
Then the author sought further to explore the molecular mechanism that leads to mitochondrial dysfunction and ultimately cell death upon ME3 depletion.
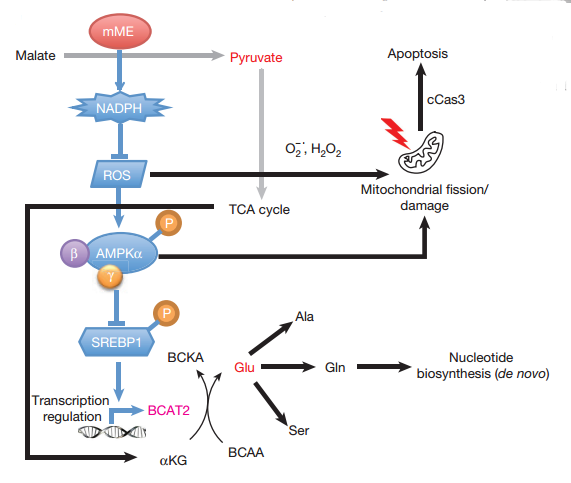
Fig2. Molecular mechanism on how ME deficient induce cell death.
As mentioned before ME3 depletion in ME2-null cells would lead in ROS accumulation, correlating with NADPH and Glutathione reduction. High intracellular ROS will induce AMPK, which is the indicator of metabolic stress. One of the known targets regulated by AMPK is SREBP1c, which is the binding site in the BCAT2 promoter. Activation of SREBP1 by mTORC1 leads to nuclear translocation of SREBP1 and subsequent transcription of its target genes. AMPK inhibits nuclear translocation of SREBP1 by phosphorylation and thereby suppresses BCAT2 expression. Besides STEBP1 could also target acetyl CoA carboxylase (ACC) and fatty acid synthase (FAS), Thus AMPK could suppresses ACC and FAS indirectly, too.
The exocrine pancreas has one of the highest BCAT2 activity of all tissues. BCAT2 is a mitochondrial isoform required for the first step in branched-chain amino acids (BCAA) catabolism, where the amino group of BCAAs is transferred to α-KG to generate Glu. Since the inhibition of BCAT2 in ME3 depletion in ME2-null cells, assimilation of BCAAs decrease, thereby BCAAs accumulate inside the cytosol. An increase in BCAAs in the circulation is an indication of high protein turnover and muscle breakdown, which are hallmarks of cancer cachexia seen in many patients with PDAC. ME3 depletion in ME2-null cells inhibited amino group transfer from BCAA to α-KG, but the BCAA flux did not change in TCA cycle flux. de novo synthesis of nucleotides required ammonia, which could be contribute from Gln. Additionally, a mixture of nucleotides could rescue the cell proliferation of ME3 depletion- ME2-null cells. Thus the author indicating that BCAA-derived nucleotide precursors are rate limiting to cell viability.
In summary, the author concluded, malic enzyme depletion resulting in (i) ROS-mediated activation of the AMPK pathway causing a decrease in BCAA breakdown and thereby leading to a decrease in de novo nucleotide biosynthesis, and (ii) ROS mediated mitochondrial damage leading to activation of apoptotic pathways.
This study provides an effort to expand therapeutic strategies beyond oncogenic targets to those not directly linked to cancer pathogenesis, and have identified a collateral lethal vulnerability in PDAC that can be targeted pharmacologically in genotype-defined patient populations.
Reference
P Dey et al. Nature 542 (7639), 119-123. 2017 Jan 18.