G-protein coupled receptors (GPCRs)
SubCategories
- Interleukin Subfamily
- GPCRs Class A
- GPCRs Class B
- GPCRs Class C
- GPCRs Class F
- GPCRs Taste/Vomeronasal Receptors
- Physiological Functions of GPCRs
- How Do GPCRs Work?
- Dysregulation of GPCRs
- GPCRs Desensitization
- GPCRs Diseases
- G-protein coupled receptor (GPCR) pathways
- GPCRs Subfamily
- G-protein Signaling
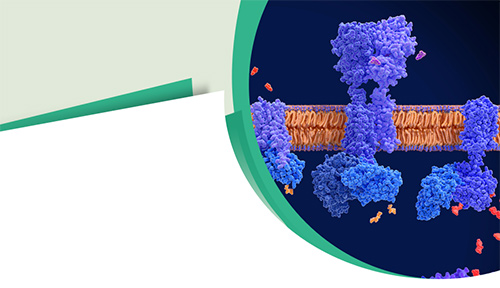
GPCRs or G protein coupled receptors are a group of cell surface receptors, for relaying external signals into cells and triggering various physiological reactions in the body such as vision taste and smell perception, immune responses and nerve signal transmission These receptors are involved in a range of important biological functions and processes. GPCRs are known to be the most varied family of membrane receptors found in the genome with approximately 800 to 1,000 members identified so far. They are also targets for drug development due to their presence and impact, on signaling pathways; about 40 percent of existing medications target GPCRs.
What is the Structure of GPCRs?
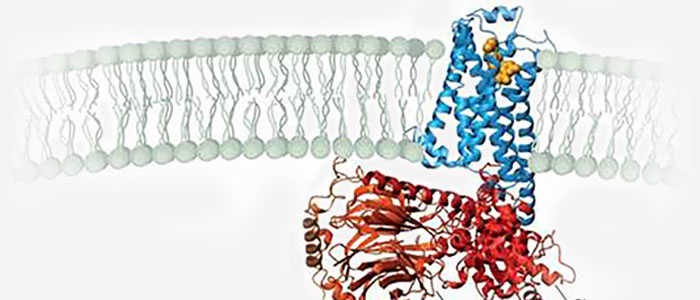
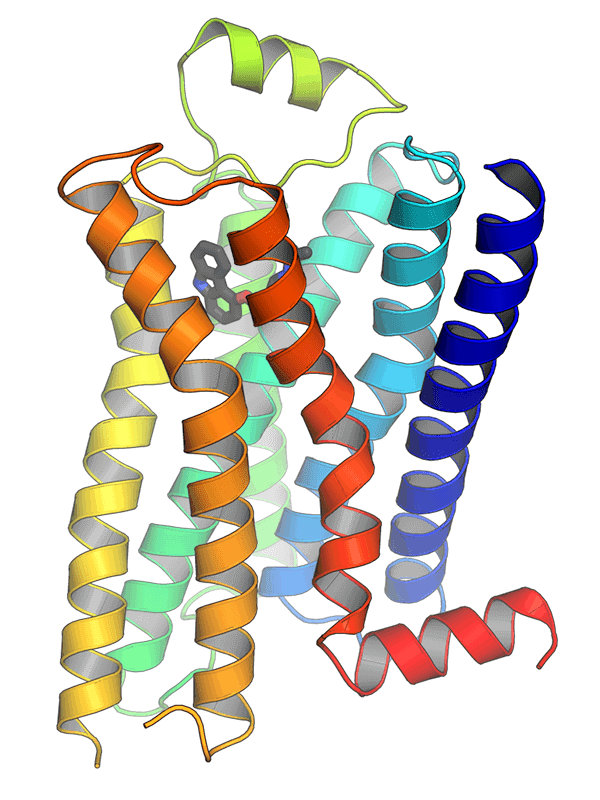
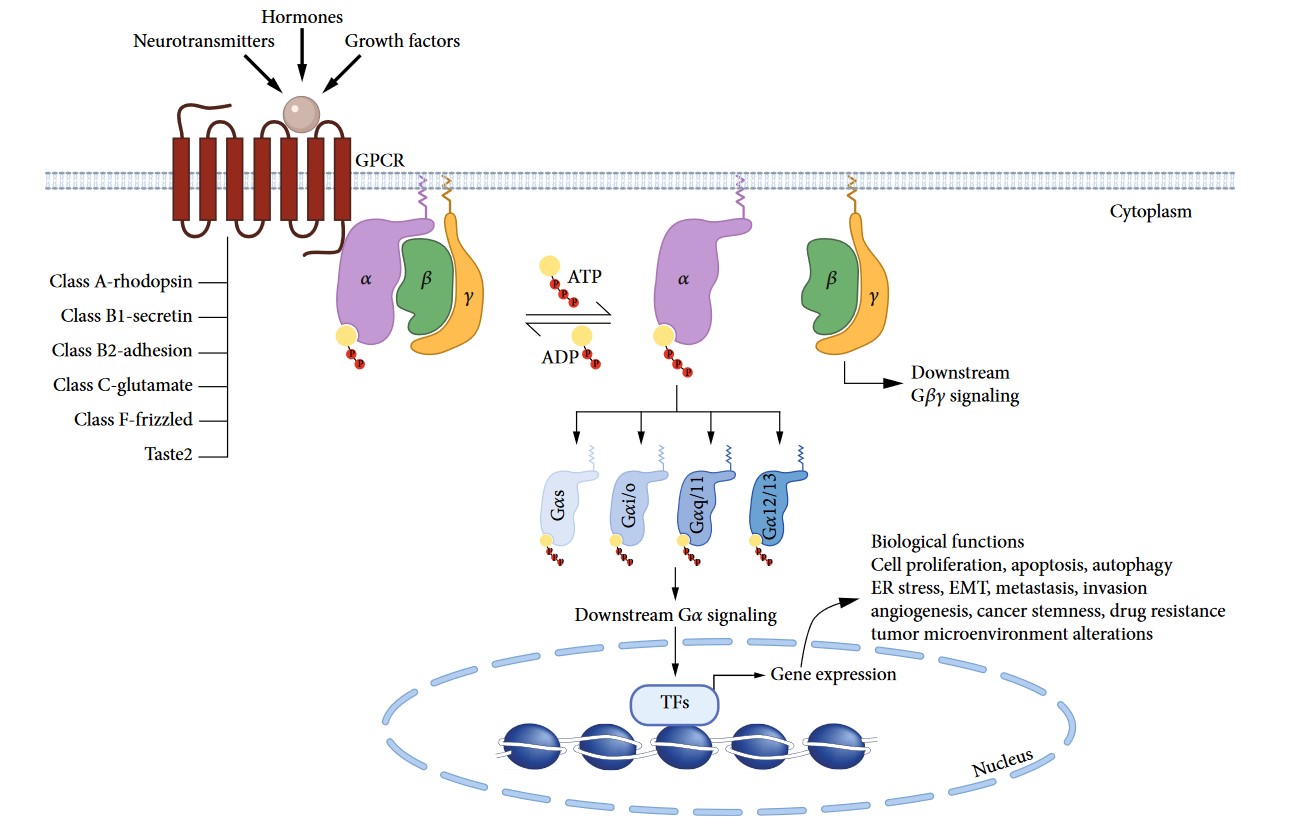
The structure of GPCRs is fundamental to their function in signal transduction. All GPCRs share a common architecture of seven transmembrane α-helices that span the plasma membrane. These helices are connected by three extracellular loops and three intracellular loops. The extracellular loops and the N-terminus are involved in ligand binding, while the intracellular loops and the C-terminus are critical for interaction with G-proteins and other intracellular signaling molecules.
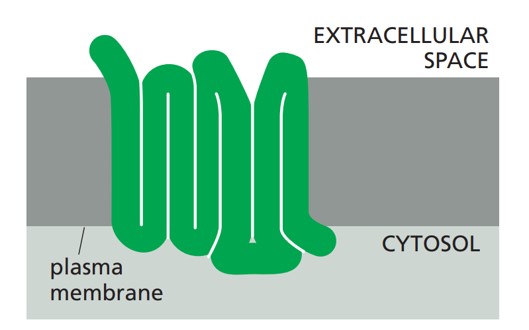
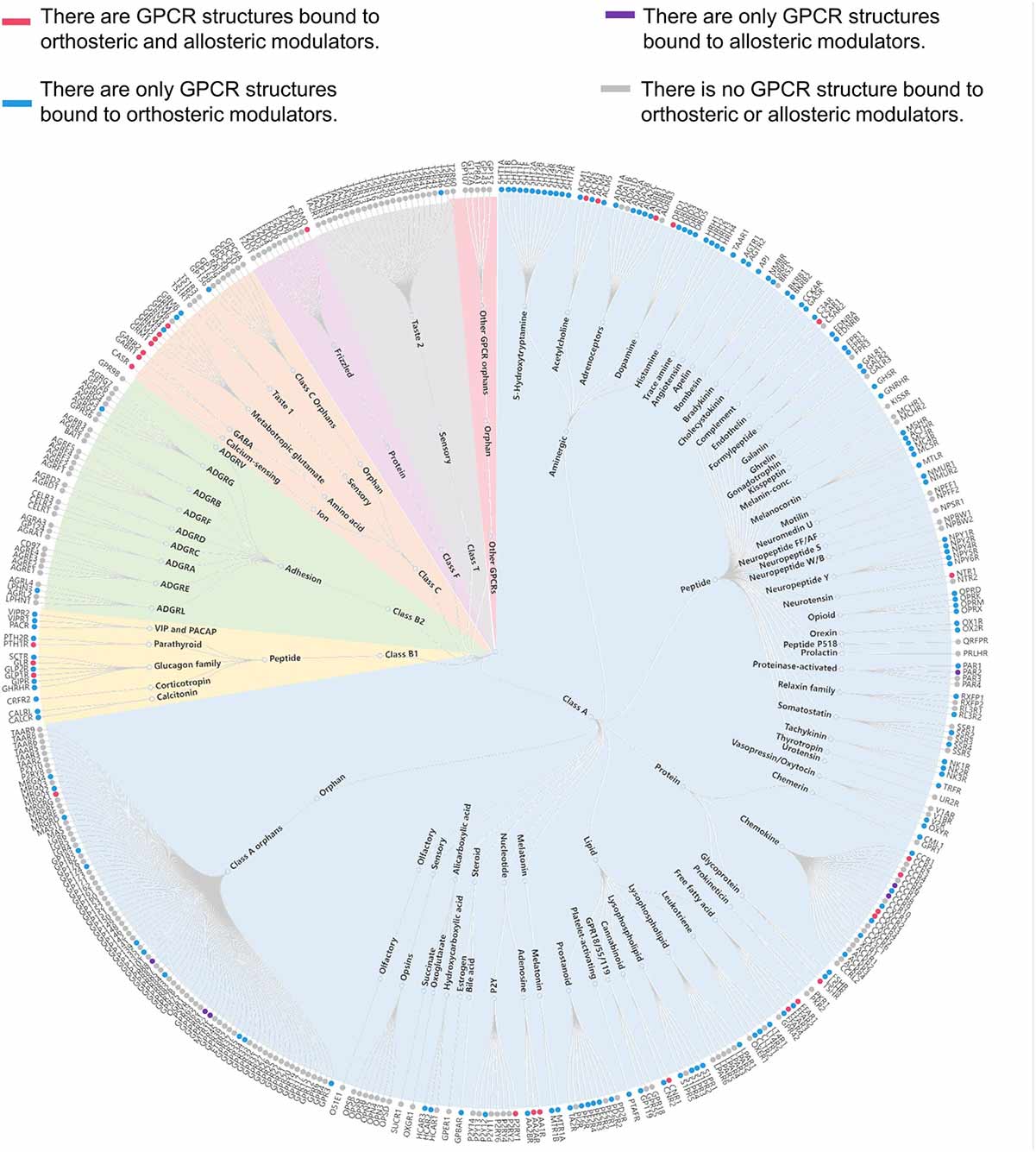
Classifications of GPCRs
GPCRs can be classified into several families or classes based on their sequence homology, structural features, and ligand-binding properties. The most widely accepted classification system groups GPCRs into five major families: Class A (Rhodopsin-like), Class B (Secretin-like), Class C (Metabotropic Glutamate-like), Class F (Frizzled/Smoothened), and Taste/Vomeronasal receptors. There are also orphan receptors, which are GPCRs that have not yet been matched with their endogenous ligands.
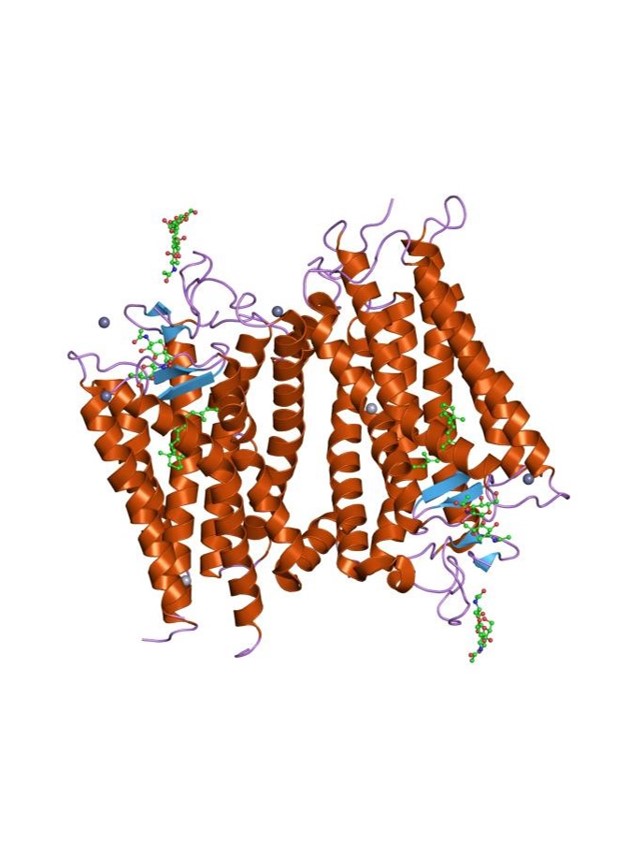
How Do GPCRs Work?
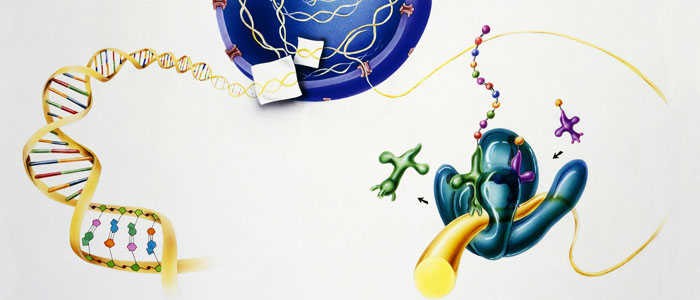
The signaling mechanism of GPCRs is initiated when an extracellular ligand binds to the receptor, inducing a conformational change in the receptor that allows it to interact with and activate an intracellular G-protein. Key steps include:
- Ligand Binding
- Activation of G-Proteins
- Effector Activation
- Signal Termination and Receptor Desensitization
Related Links
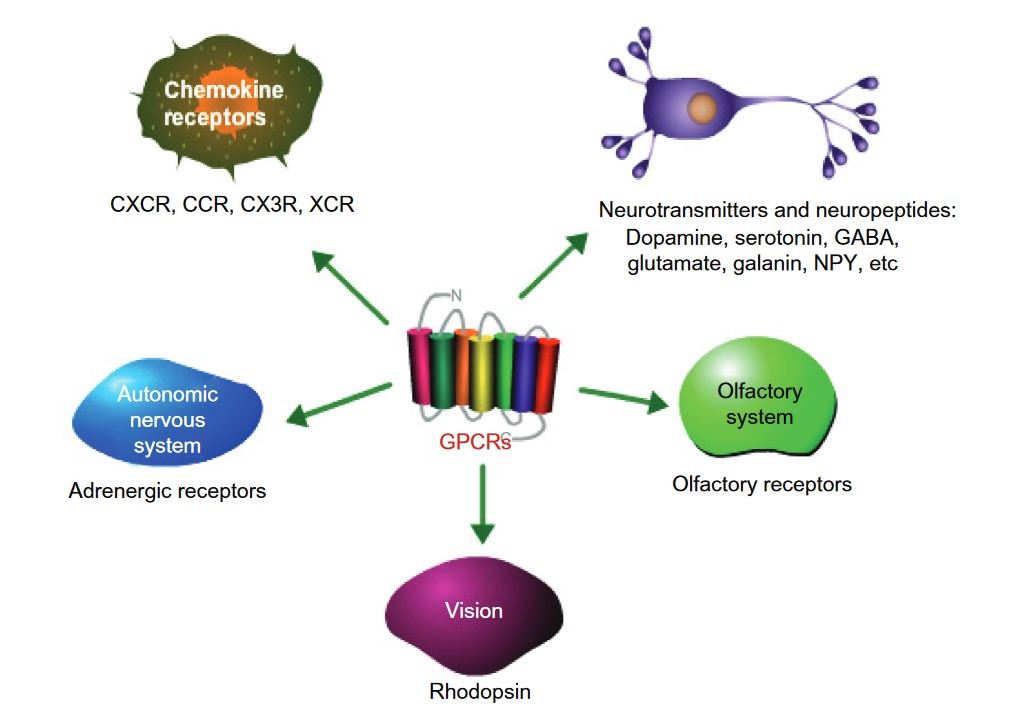
Physiological Functions of GPCRs
GPCRs have a wide range of important roles in the body, helping to regulate many essential biological processes. These receptors are key players in the regulation of physiological functions across various systems, including the nervous, endocrine, cardiovascular, immune, and sensory systems.
Related Links
Dysregulation of GPCRs
Dysregulation of GPCR signaling can lead to a variety of diseases, including cancer, cardiovascular disease, neurological disorders, and metabolic conditions.
Related Links
GPCRs Agonists and Antagonists
GPCRs are influenced by two types of molecules: agonists and antagonists. Agonists bind to GPCRs and activate them, effectively "switching on" the receptor to trigger specific cellular responses. These molecules play a key role in regulating physiological processes such as mood, heart rate, and immune system activity, with hormones and neurotransmitters often acting as natural agonists. Antagonists , by contrast, also bind to GPCRs but work differently. Instead of activating the receptor, they block it from being triggered by other molecules. This "blocking" action can prevent harmful overactivation of GPCRs, which is often associated with diseases or disorders. By either enhancing or inhibiting GPCR activity, agonists and antagonists are essential tools in drug development. They offer a foundation for therapies aimed at treating a wide range of medical conditions with precision and effectiveness.
Tab. 1: Typical GPCRs and their corresponding agonists and antagonists.
| Receptors | Agonists | Antagonists | |
|---|---|---|---|
|
5-HT1 (subtypes 1A, 1B, 1D etc.) |
Serotonin |
/ |
|
|
5-HT2 (subtypes 2A, 2B, etc.) |
|||
|
Dopamine receptors |
Dopamine |
Haloperidol |
|
|
α1-adrenergic receptors |
Norepinephrine |
Prazosin |
|
|
/ |
|||
|
β1-adrenergic receptors |
Propranolol |
||
|
β2-adrenergic receptors |
|||
|
Thyroid-stimulating hormone (TSH) |
Small-molecule inhibitors |
||
|
Glucagon |
LY2409021 |
||
|
Gonadotropin-releasing hormone receptors |
Gonadotropin-releasing hormone (GnRH) |
Cetrorelix |
|
|
Oxytocin |
Atosiban |
||
|
Angiotensin II receptor |
Angiotensin II |
Losartan |
|
|
Losartan |
|||
|
Chemokine receptors |
CXCR4 (for SDF-1) |
Chemokines (RANTES, MIP-1) |
/ |
|
CCR5 |
Maraviroc |
||
|
Histamine receptors |
Histamine |
Diphenhydramine |
|
|
Ranitidine |
|||
|
Ciproxifan |
|||
|
JNJ 7777120 |
|||
|
Purinergic receptors |
P1 (A1, A2A, A2B, A3) |
Adenosine |
Caffeine (A1, A2A) |
|
ATP |
|||
|
Rhodopsin GPCR (Opsins) |
Light (photons) |
/ |
|
|
Olfactory receptors (ORs) |
Odor molecules |
/ |
|
|
Taste GPCRs |
Sugars |
Lactisole |
|
|
Bitter compounds (quinine, etc.) |
/ |
||
|
GLP-1 |
Exendin (9-39) |
||
|
Free fatty acid receptors |
Fatty acids |
GW1100 |
|
|
/ |
|||
|
Frizzled receptors |
Wnt proteins |
Wnt inhibitors |
|
|
Sonic Hedgehog (Shh) |
Vismodegib |
||
|
Opioid receptors |
Morphine |
Naloxone |
|
|
Cannabinoid receptors |
THC |
Rimonabant |
|
|
AM630 |
|||
|
Follicle-stimulating hormone (FSH) |
Small-molecule inhibitors |
||
|
Luteinizing hormone (LH) |
Ganirelix (indirect antagonist by blocking GnRH) |
||
GPCRs as Drug Targets
Because of their extensive involvement in a wide range of physiological processes and diseases, GPCRs have become one of the most important target classes. About 40% of all prescription drugs in the world target GPCRs, highlighting their importance in pharmacotherapy. But even with so many GPCRs, most commercial drugs target only a small subset of them, leaving many potential targets unexplored.
Related Links