



Apoptosis Signal Pathway Overview

🧪 TNF-24H
Source: E.coli
Species: Human
Tag: Non
Conjugation:
Protein Length:

🧪 NGF-05H
Source: CHO
Species: Human
Tag: Non
Conjugation:
Protein Length: 122-239 aa

🧪 IL3-120H
Source: HEK293
Species: Human
Tag: Non
Conjugation:
Protein Length:

🧪 IL3RA-121H
Source: HEK293
Species: Human
Tag: Fc
Conjugation:
Protein Length: Thr19-Arg305

🧪 GZMB-296H
Source: HEK293
Species: Human
Tag: His
Conjugation:
Protein Length: 1-247 aa

🧪 DIABLO-869H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: Ala56-Asp239

🧪 Gzmb-942M
Source: Insect Cells
Species: Mouse
Tag: Non
Conjugation:
Protein Length:

🧪 RAF1-1123H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 50-132 a.a.

🧪 TNF-412H
Source: HEK293
Species: Human
Tag: Flag
Conjugation:
Protein Length: 85-233;18-111 a.a.
 Full L.
Full L.
🧪 PARP1-650H
Source: Insect Cells
Species: Human
Tag: His
Conjugation:
Protein Length: 1-1014 a.a.

🧪 Ctsa-908M
Source: HEK293
Species: Mouse
Tag: His
Conjugation:
Protein Length: Met1-Tyr474

🧪 FAS-3154H
Source: HEK293
Species: Human
Tag: His
Conjugation:
Protein Length: Met1-Asn173

🧪 IL3RA-3178H
Source: HEK293
Species: Human
Tag: His
Conjugation:
Protein Length: 1-305 a.a.

🧪 Fas-3268M
Source: HEK293
Species: Mouse
Tag: His
Conjugation:
Protein Length: Met1-Arg169

🧪 IL3RA-3248H
Source: HEK293
Species: Human
Tag: Fc&His
Conjugation:
Protein Length: 1-305 aa

Background of Apoptosis Signal Pathway
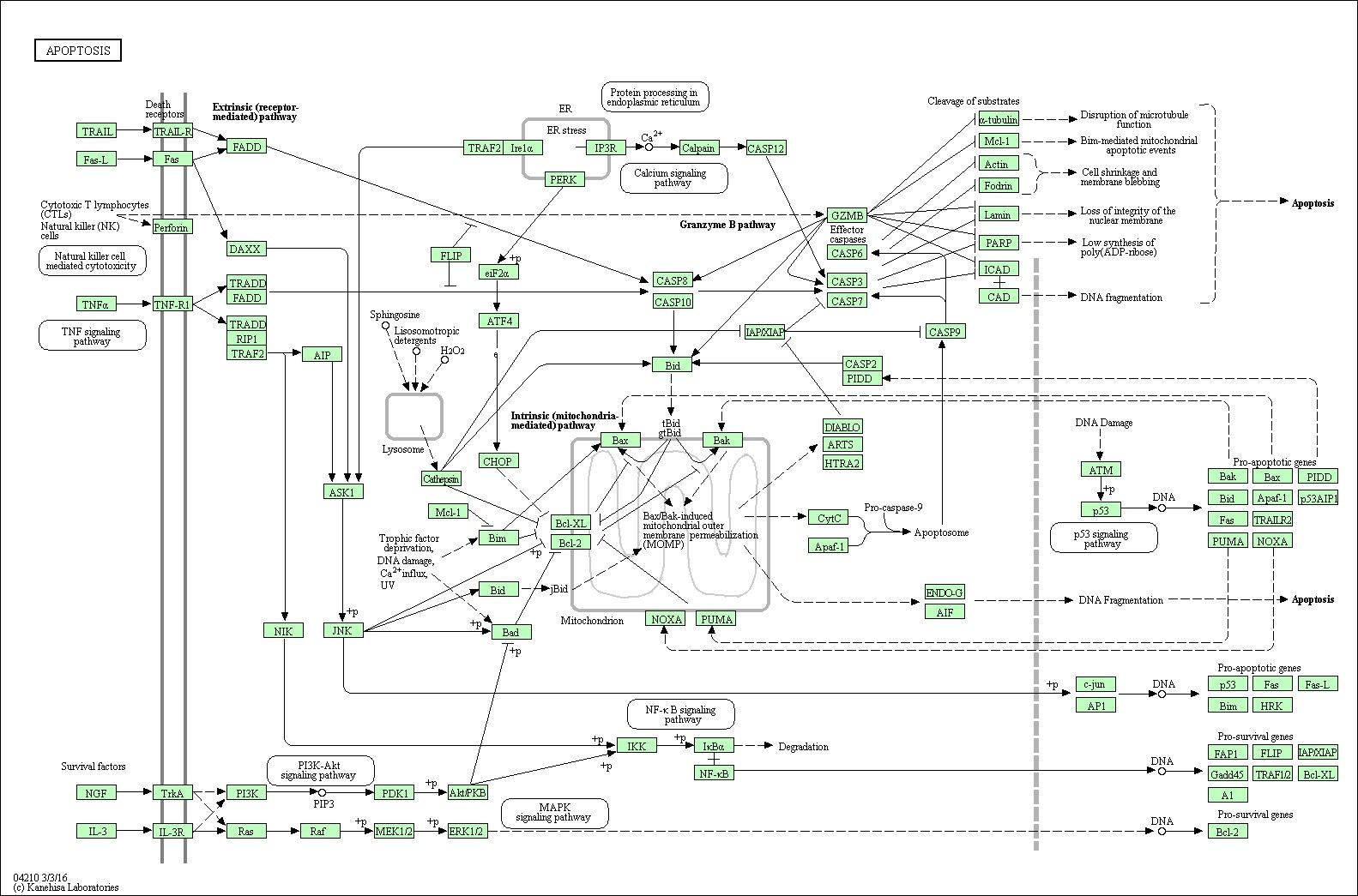
Apoptosis is a kind of programmed cell death (PCD) plays a fundamental role in multicellular organism development and tissue homeostasis. We might take it as a regulated cellular suicide mechanism. Abnormal regulation of this process is associated with a wide variety of human diseases, including immunological and developmental disorders, neurodegeneration, and cancer. In contrast to necrosis, which is a form of traumatic cell death that results from acute cellular injury, apoptosis is a highly regulated and controlled process that confers advantages during an organism's lifecycle. Unlike necrosis, apoptosis produces cell fragments called apoptotic bodies that phagocytic cells are able to engulf and quickly remove before the contents of the cell can spill out onto surrounding cells and cause damage.
The initiation of apoptosis is tightly regulated by activation mechanisms, and once apoptosis has begun, it inevitably leads to the death of the cell. There are at least three different kinds of pathways participate in apoptosis initiation: death receptor signaling pathway, mitochondrial pathway and endoplasmic reticulum(ER) stress pathway. As a pathway is more or less sequential in nature, removing or modifying one component leads to an effect in another. Inhibition of apoptosis can result in a number of cancers, autoimmune diseases, inflammatory diseases, and viral infections. On the other hand, excess apoptosis can lead to neurodegenerative diseases, hematologic diseases, and tissue damage with a cell biology phenomenon that is loss of control of the cell death.

Fig1. Schematic representation of apoptotic events. (Susan Elmore, 2007)
Mechanisms to Initiate Apoptosis
Two typical theories of the direct initiation of apoptotic mechanisms have been raised: the TNF-induced (tumour necrosis factor) model and the Fas-Fasligand mediated model. TNF-alpha is a cytokine produced mainly by activated macrophages, and is the major extrinsic mediator of apoptosis. The binding of TNF-alpha to TNFR1 was able to initiate the pathway leads to caspase activation via the intermediate membrane proteins TNF receptor-associated death domain (TRADD) or Fas-associated death domain protein (FADD). The First apoptosis signal (Fas, also known as Apo-1 or CD95) receptor binds the Fas ligand (FasL), a transmembrane protein part of the TNF family. The interaction between Fas and FasL results in the formation of the death-inducing signaling complex (DISC), which contains the FADD, caspase-8 and caspase-10. In some types of cells (type I), processed caspase-8 directly activates other members of the caspase family, and triggers the execution of apoptosis of the cell. In other types of cells (type II), the Fas-DISC starts a feedback loop that spirals into increasing release of proapoptotic factors from mitochondria and the amplified activation of caspase-8. Activation of caspases ensure that the cellular components are degraded in a controlled manner, carrying out cell death with minimal effect to surrounding tissues.
Key Organelles of Apoptosis Signal Pathway
The mitochondria are essential to multicellular creatures. The mitochondria pathway of apoptosis functions is in response to various types of intracellular stress including growth factor withdrawal, DNA damage, unfolding stresses in the ER and death receptor stimulation. Apoptotic proteins such as BCL-2 family proteins target mitochondria and affect them in different ways. They might lead to mitochondrial swelling and increase the permeability of the mitochondrial membrane, as a result, the apoptotic effectors will leak out. Also, the nitric oxide is able to induce apoptosis by helping to dissipate the membrane potential of mitochondria and therefore make it more permeable. Mitochondrial proteins known as second mitochondria derived activator of caspases (Smac) are released into the cell's cytosol, following the increase in permeability of the mitochondria membranes. Smac binds to proteins that inhibit apoptosis (IAPs), thereby deactivating them, and preventing the IAPs from arresting the process and therefore allowing apoptosis to proceed. IAP also normally suppresses the activity of a group of cysteine proteases called caspases, which carry out the degradation of the cell. Therefore, the actual degradation enzymes can be seen to be indirectly regulated by mitochondrial permeability.
The ER is the organelle responsible for synthesis and folding of secreted and membranous protein and lipid biosynthesis. The ER stress response constitutes a cellular process that is triggered bya variety of conditions that disturb folding of proteins in the ER. The ER stress response is mediated by three sensors located at the ER membrane: ERN1, ATF6and PERK/EIF2A3K. Accumulation of unfolded protein recruits BiP/HSP70 to the ER lumen and its dissociation from ERN1, ATF6 and PERK/EIF2A3K leads to their activation. ERN1 can recruit TRAF2and ASK1, leading to downstream activation of JNKand p38 MAPK. Activated JNK translocates to the mitochondrial membrane and promotes activation of Bimand inhibition of Bcl-2. Baxand Bakbind to and activate IRE1α and induce release of Ca2+from the ER to mitochondria. Then PERK phosphorylates elF2α and attenuates protein translation. Activated ATF6 translocates to the Golgi and cleaved by the proteases, S1P and S2P. The cleaved ATF6 fragment forms an active transcriptional factor that mediates expression of several components important for protein folding, degradation, and ER expansion.

Fig2. Mechanisms leading to deregulation of apoptosis. (Giuseppa Pistritto, 2016)
Core Components of Apoptosis Signal Pathway
Many factors involved in the regulation of apoptosis. Some factors like Fas, TNFαR, DR3, DR4, DR5 receptors, p53proteins and caspases promote apoptosis via receptor oligomerization, while some members of the Bcl-2 family like Bcl-2, Bcl-x1, Bcl-w, Mcl-1 proteins inhibit apoptosis.While members like Bax, Bak, Bad, Bid and Bim could promote apoptosis. Bcl-2 proteins are able to inhibit or promote apoptosis by direct action on MAC. Of all the three pathways above, caspases play an essential role in them. They finally activate caspases which leading to apoptosis. But there also exists a caspase-independent apoptotic pathway that is mediated by apoptosis-inducing factor (AIF).
Caspases are proteins that are highly conserved, cysteine-dependent aspartate-specific proteases. There are two types of caspases: initiator caspases, caspase -2, -8, -9, -10, -11, -12, and effector caspases, caspase -3, -6,-7. The activation of initiator caspases requires binding to specific oligomericactivator protein. Effector caspases are then activated by these active initiator caspases through proteolytic cleavage. The active effector caspases then proteolytically degrade a host of intracellular proteins to carry out the cell death program.
Case Study
Case Study 1: Recombinant Human FAS (FAS-281H)
Serum soluble Fas (sFas) levels are associated with erythropoietin (Epo) hyporesponsiveness in patients with chronic kidney disease (CKD). Whether sFas could predict the need for erythropoiesis-stimulating agent (ESA) usage and its influence in erythropoiesis remain unclear. The researchers evaluated the relation between sFas and ESA therapy in patients with CKD with anemia and its effect on erythropoiesis in vitro. First, they performed a retrospective cohort study with 77 anemic patients with nondialysis CKD. They performed in vitro experiments to investigate whether sFas could interfere with the behavior of hematopoietic stem cells (HSCs). HSCs were isolated from umbilical cord blood and incubated with recombinant sFas protein in a dose-dependent manner. Serum sFas positively correlated with Epo levels (r = 0.30, P = 0.001) but negatively with hemoglobin (r = -0.55, P < 0.001) and glomerular filtration rate (r = -0.58, P < 0.001) in patients with CKD at baseline. Elevated sFas serum levels (4,316 ± 897 vs. 2,776 ± 749, P < 0.001) with lower estimated glomerular filtration rate (26.2 ± 10.1 vs. 33.5 ± 14.3, P = 0.01) and reduced hemoglobin concentration (11.1 ± 0.9 vs. 12.5 ± 1.2, P < 0.001) were identified in patients who required ESA therapy compared with patients with non-ESA. Afterward, they detected that the sFas level was slight correlated with a necessity of ESA therapy in patients with nondialysis CKD and anemia. In vitro assays demonstrated that the erythroid progenitor cell frequency negatively correlated with sFas concentration (r = -0.72, P < 0.001).

Fig1. Analyze of the human recombinant soluble Fas (sFas) influence on in vitro erythropoiesis. (Daniela Mendes Chiloff, 2020)
Case Study 2: Recombinant Human RIPK1 (RIPK1-7018H)
Genomic instability promotes colon carcinogenesis by inducing genetic mutations, but not all genes affected by this process have been identified. The researchers investigated whether genomic instability in human colorectal cancer (CRC) cells produces mutations in the hepatocyte growth factor (HGF) gene. They genotyped human colon tumor tissues and adjacent nontumor tissues collected from 78 patients University of Pittsburgh Health Sciences and Veterans Hospital, along with 40 human CRC and adjacent nontumor tissues in a commercial microarray. They used cellular, biochemical, and molecular biological techniques to investigate the factors that alter HGF signaling in colon cancer cells and its effects on cell proliferation and survival. The results showed that HGF signaling via MET reduced levels of the receptor-interacting serine-threonine kinase 1 (RIPK1), a mediator of necroptosis, in CRC cells. High levels of HGF protein in tumor tissues correlated with lower levels of RIPK1 and shorter survival times of patients.

Fig2. Met activation by HGF treatment down regulates RIPK-1. (Danushka Seneviratne, 2015)
Case Study 3: Recombinant Human IRE1 protein (ERN1-1124H)
IRE1α is constitutively active in several cancers and can contribute to cancer progression. Activated IRE1α cleaves XBP1 mRNA, a key step in production of the transcription factor XBP1s. In addition, IRE1α cleaves select mRNAs through regulated IRE1α-dependent decay (RIDD). Accumulating evidence implicates IRE1α in the regulation of lipid metabolism. However, the roles of XBP1s and RIDD in this process remain ill-defined. In this study, transcriptome and lipidome profiling of triple negative breast cancer cells subjected to pharmacological inhibition of IRE1α reveals changes in lipid metabolism genes associated with accumulation of triacylglycerols (TAGs). The researchers identify DGAT2 mRNA, encoding the rate-limiting enzyme in TAG biosynthesis, as a RIDD target. Inhibition of IRE1α, leads to DGAT2-dependent accumulation of TAGs in lipid droplets and sensitizes cells to nutritional stress, which is rescued by treatment with the DGAT2 inhibitor PF-06424439.

Fig3. Wild type (WT) DGAT2, c.260G>A DGAT2 mutant (MUT DGAT2) were incubated with recombinant hIRE1α in the presence or absence of 20 μM MKC8866. (Aitor Almanza, 2022)
Related Products
Apoptosis, also known as Programmed Cell Death (PCD), is an autonomous, orderly process of cell death controlled by the cell's internal programs. Apoptosis plays an important role in the development, maintenance of tissue homeostasis and immune defense of multicellular organisms. Creative BioMart can provide a list of core protein products of the Apoptosis Signal Pathway to help you with your researches. Please feel free to contact us if you’re interested.