



Mitochondrial Control of Apoptosis
🧪 RAF1-1123H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 50-132 a.a.

🧪 FAS-3154H
Source: HEK293
Species: Human
Tag: His
Conjugation:
Protein Length: Met1-Asn173

🧪 Fas-3268M
Source: HEK293
Species: Mouse
Tag: His
Conjugation:
Protein Length: Met1-Arg169

🧪 Bid-4029M
Source: E.coli
Species: Mouse
Tag: GST&His
Conjugation:
Protein Length: Met1-Asp195
🧪 Fas-4096R
Source: HEK293
Species: Rat
Tag: His
Conjugation:
Protein Length: Met1-Lys170

🧪 PTEN-1619H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-403 a.a.
.jpg)
🧪 BCL2-26734TH
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-451 a.a.
🧪 ATM-9987H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1332-1479a.a.
🧪 DAXX-11834H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-349a.a.

🧪 ENDOG-12448H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 63-297a.a.

🧪 FAS-12751H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: C-term-180a.a.


🧪 MDM2-350H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 1-118 a.a.

🧪 PTEN-2042H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 204-403aa

🧪 RAF1-2161H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: C-term-300aa

🧪 FAS-281H
Source: Human Cells
Species: Human
Tag: GST&His
Conjugation:
Protein Length: 1-173 a.a.

Background of Mitochondrial Control of Apoptosis
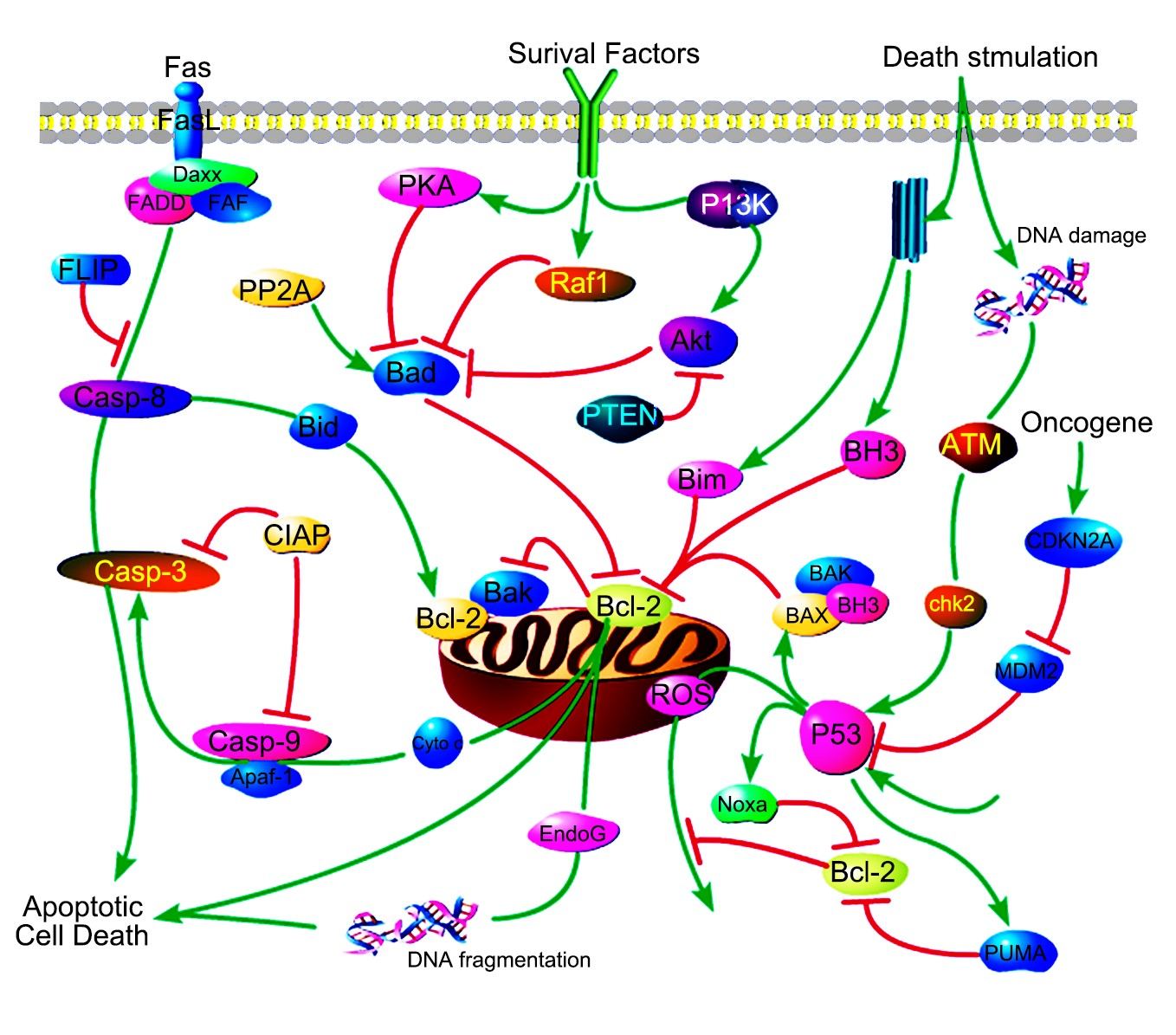
What is Mitochondrial Control of Apoptosis?
Mitochondrial apoptosis, often referred to simply as apoptosis, is a form of programmed cell death that plays a crucial role in maintaining the health of multicellular organisms. It is a highly regulated process that involves a series of biochemical events leading to the systematic dismantling of a cell. The mitochondria, often described as the "powerhouses of the cell" due to their role in energy production, are central to the initiation and execution of apoptosis.
Apoptosis is a form of programmed cell death (PCD) that is distinct from necrosis, which is a form of traumatic cell death. It is essential during development for processes such as tissue sculpting and the removal of superfluous cells. Apoptosis helps maintain tissue homeostasis by eliminating damaged or infected cells.
Mitochondria are central to the intrinsic pathway of apoptosis. Under certain stimuli, such as DNA damage or growth factor deprivation, the mitochondrial outer membrane permeabilizes, releasing cytochrome c into the cytosol. Along with cytochrome c, other apoptogenic factors like SMAC/DIABLO and apoptosis-inducing factor (AIF) are also released.
Caspases are a family of cysteine proteases triggered by apoptosis signaling. The culmination ofcaspasecascade is the cleavage of a number of proteins in the cell, followed by cell disassembly,cell death, and, ultimately, the phagocytosis and removal of the cell debris. The caspasecascade can be activated by two distinct routes: one from cell surface and the other from mitochondria. The mitochondrial pathway of apoptosis, also called theintrinsic pathway, is the most common mechanism of apoptosis in vertebrates.
Core Components of Mitochondrial Control of Apoptosis
Cytochrome c is an essential component of the mitochondrial electron transport chain. It is translated as apocytochrome c and then translocated into the mitochondrial intermembrane space. In the unstimulated situation, cytochrome c is trapped at the inner mitochondrial membrane by interaction with the lipid cardiolipin. Upon receipt of the appropriate stimulation, one consequence is an increase in the permeability of the outer mitochondrial membrane, which allows escape of the cytochrome c from the intermembrane space, via formation of a channel, called the mitochondrial apoptosis-induced channel (MAC) in the outer mitochondrial membrane. In a further step of apoptosis, Cytochrome c will escape, then binds with Apoptotic protease activating factor - 1 (Apaf-1) and ATP, which then bind to pro-caspase-9 to create a protein complex termed apoptosome.

Fig1. Cytochrome-c released from mitochondrial. (Sen Zhang, 2022)
The apoptosome requires ATP for its formation and is able to cleave and activate procaspase-3, which is an effector caspase. The adaptor protein Apaf1 appears to play a major structural role in this assembly. Apaf1 contains WD motifs for interaction with cytochrome c and a CARD motif, which directs binding to the CARD motifs of procaspase-9 and procaspase-3. Structural studies on the apoptosome by electron microscopy have revealed a wheel shaped heptameric complex, with the CARD domains of Apaf-1 located at the central hub and the WD40 repeats at the extended spokes. The location of procaspase 9 in this complex is still open as is the mechanism of caspase 9 activation. The activation of caspase-9 by apoptosome formation sets in motion a cascade of caspase activation events. At the top of this cascade is caspase 3, which cleaves other downstream effector procaspases (caspases-2, -6, -8 and -10) or apoptotic substrates containing the recognition motif DXXD. Activation of this hierarchically structured cascade leads to proteolyis of multiple substrates, and the cell is committed to death. The cellular infrastructure is destroyed, and changes at the plasma membrane are triggered that promote engulfment by phagocytes.
In addition to cytochrome c, other soluble proteins contained in the inter-membrane space of mitochondria are released through the outer membrane and participate in the organized destruction of the cell, too. Among these proteins are the apoptosis inducing factor (AIF), endonuclease G, and the second mitochondria derived activator of caspase (Smac)/Diablo proteins. The AIF protein hasbeen shown to be responsible for triggering a pathway of programmed cell deaththat is distinct from the caspase pathways. Endonuclease G translocates during apoptosis from the mitochondrion to the nucleus, where it cleaves chromatin DNA intonucleosomal fragments about 50Kb independently of caspases. Smac/Diablo is also released from the mitochondria along with cytochrome c during apoptosis, and it functions to promote caspase activation by inhibiting Inhibitor of apoptosis family proteins (IAPs). Smacs prevent the IAPs from arresting the apoptotic process and therefore allowing apoptosis to proceed. IAPs also suppress the activity of caspases, which carry out the degradation of the cell.
Bcl-2-family proteins can regulate the amount of releasable Ca2+ thereby helping to determine whether mitochondria undergo the permeability transition and hence death. Apoptosis regulators like BCL-2 and Bcl-2 homology (BH) are family of evolutionarily related proteins. These proteins govern mitochondrial outer membrane permeabilization (MOMP) and can be either pro-apoptotic (Bax, Bad, Bakand Bokamong others) or anti-apoptotic (including Bcl-2 proper, Bcl-xL, and Bcl-w, among an assortment of others). Mitochondria can release intermembrane space (IMS) proteins through two different mechanisms: through a surgically precise permeabilization of the outer membrane mediated by Bcl-2 family members; or through induction of the mitochondrial permeability transition (PT), occurring mainly in response to the release of Ca2+ from endoplasmic reticulum (ER) stores. Bcl-2-family proteins also regulate this pathway through their localization at the ER.

Fig2. BH domain organization of selected members of the BCL-2 family.
Case Study
Case Study 1: Human Prekallikrein Activator (PKA) Reference standard (PKA-9H)
Ryanodine receptor (RyR), a calcium-release channel located in the sarcoplasmic reticulum membrane of muscles, is the target of insecticides used against a wide range of agricultural pests. Mammalian RyRs have been shown to be under the regulatory control of several kinases and phosphatases, but little is known about the regulation of insect RyRs by phosphorylation. Here the researchers present the crystal structures of wild-type and phospho-mimetic RyR Repeat34 domain containing PKA phosphorylation sites from diamondback moth (DBM), a major lepidopteran pest of cruciferous vegetables. Using tandem mass spectrometry, they identify several PKA sites clustering in the phosphorylation loop and the newly identified α-helix. Bioinformatics analysis shows that this α-helix is only present in Lepidoptera, suggesting an insect-specific regulation. Interestingly, the specific phosphorylation pattern is temperature-dependent.

Fig1. High-temperature phosphorylation sites of DBM RyR Repeat34 measured by LC-MS/MS. (Tong Xu, 2019)
Case Study 2: Recombinant human MDM2 (MDM2-350H)
A novel class of small-molecule inhibitors of MDM2-p53 interaction with a (E)-3-benzylideneindolin-2-one scaffold was identified using an integrated virtual screening strategy that combined both pharmacophore- and structure-based approaches. The hit optimisation identified several compounds with more potent activity than the hit compound and the positive drug nutlin-3a, especially compound 1b, which exhibited both the highest binding affinity to MDM2 (Ki = 0.093 μM) and the most potent antiproliferative activity against HCT116 (wild type p53) cells (GI50 = 13.42 μM). Additionally, 1b dose-dependently inhibited tumour growth in BALB/c mice bearing CT26 colon carcinoma, with no visible sign of toxicity. In summary, compound 1b represents a novel and promising lead structure for the development of anticancer drugs as MDM2-p53 interaction disruptors.

Fig2. The proposed binding mode of compound 4b to MDM2 (generated using PyMol). (Guang-hui Zheng, 2014)
Case Study 3: Recombinant Human FAS (FAS-281H)
Serum soluble Fas (sFas) levels are associated with erythropoietin (Epo) hyporesponsiveness in patients with chronic kidney disease (CKD). Whether sFas could predict the need for erythropoiesis-stimulating agent (ESA) usage and its influence in erythropoiesis remain unclear. The researchers evaluated the relation between sFas and ESA therapy in patients with CKD with anemia and its effect on erythropoiesis in vitro. First, they performed a retrospective cohort study with 77 anemic patients with nondialysis CKD. They performed in vitro experiments to investigate whether sFas could interfere with the behavior of hematopoietic stem cells (HSCs). HSCs were isolated from umbilical cord blood and incubated with recombinant sFas protein in a dose-dependent manner. Serum sFas positively correlated with Epo levels (r = 0.30, P = 0.001) but negatively with hemoglobin (r = -0.55, P < 0.001) and glomerular filtration rate (r = -0.58, P < 0.001) in patients with CKD at baseline. Elevated sFas serum levels (4,316 ± 897 vs. 2,776 ± 749, P < 0.001) with lower estimated glomerular filtration rate (26.2 ± 10.1 vs. 33.5 ± 14.3, P = 0.01) and reduced hemoglobin concentration (11.1 ± 0.9 vs. 12.5 ± 1.2, P < 0.001) were identified in patients who required ESA therapy compared with patients with non-ESA. Afterward, they detected that the sFas level was slight correlated with a necessity of ESA therapy in patients with nondialysis CKD and anemia. In vitro assays demonstrated that the erythroid progenitor cell frequency negatively correlated with sFas concentration (r = -0.72, P < 0.001).

Fig3. Analyze of the human recombinant soluble Fas (sFas) influence on in vitro erythropoiesis. (Daniela Mendes Chiloff, 2020)
Related Products
Mitochondrial apoptosis is a complex, tightly regulated process that is fundamental to the survival and health of organisms. Understanding the molecular mechanisms underlying this process is critical for developing therapeutic strategies to combat diseases where apoptosis is a key factor. Creative BioMart can provide a list of core protein products of the apoptosis to help you with your researches. Please feel free to contact us if you’re interested.