Uncategorized Friday, 2016/11/04
Inflammatory bowel disease (IBD) is characterized by chronic intestinal inflammation and dysfunction of the epithelial barrier. The primary cause of the initiation of this disease is unclear. However, it is well accepted that inflammatory responses, most likely driven by the microbiome and defective barrier function, promote a vicious cycle that leads to chronic disease. The immune system and the microbiota mutually interact to maintain homeostasis in the intestine. However, components of the microbiota can alter this balance and promote chronic inflammation, promoting intestinal tumor development.
Bacteria can drive tumor growth, and their interaction with the immune system can further fuel cancer progression. This is especially true at mucosal interfaces where bacteria are abundant and the immune system is highly reactive.
Microbial products could promote tumor growth in two ways, directly or indirectly. Bacteria per se are not able to promote tumor initiation and growth unless they interact with the immune system, which ultimately promotes cancer. The adaptive arm of the host immune system is comprised of cells that respond selectively and specifically to infectious agents that the innate cells have collected and presented to them. Some of these responses can be protumorigenic. For example, upon contact with specific bacteria, CD4+ T cells can produce protumorigenic cytokines.
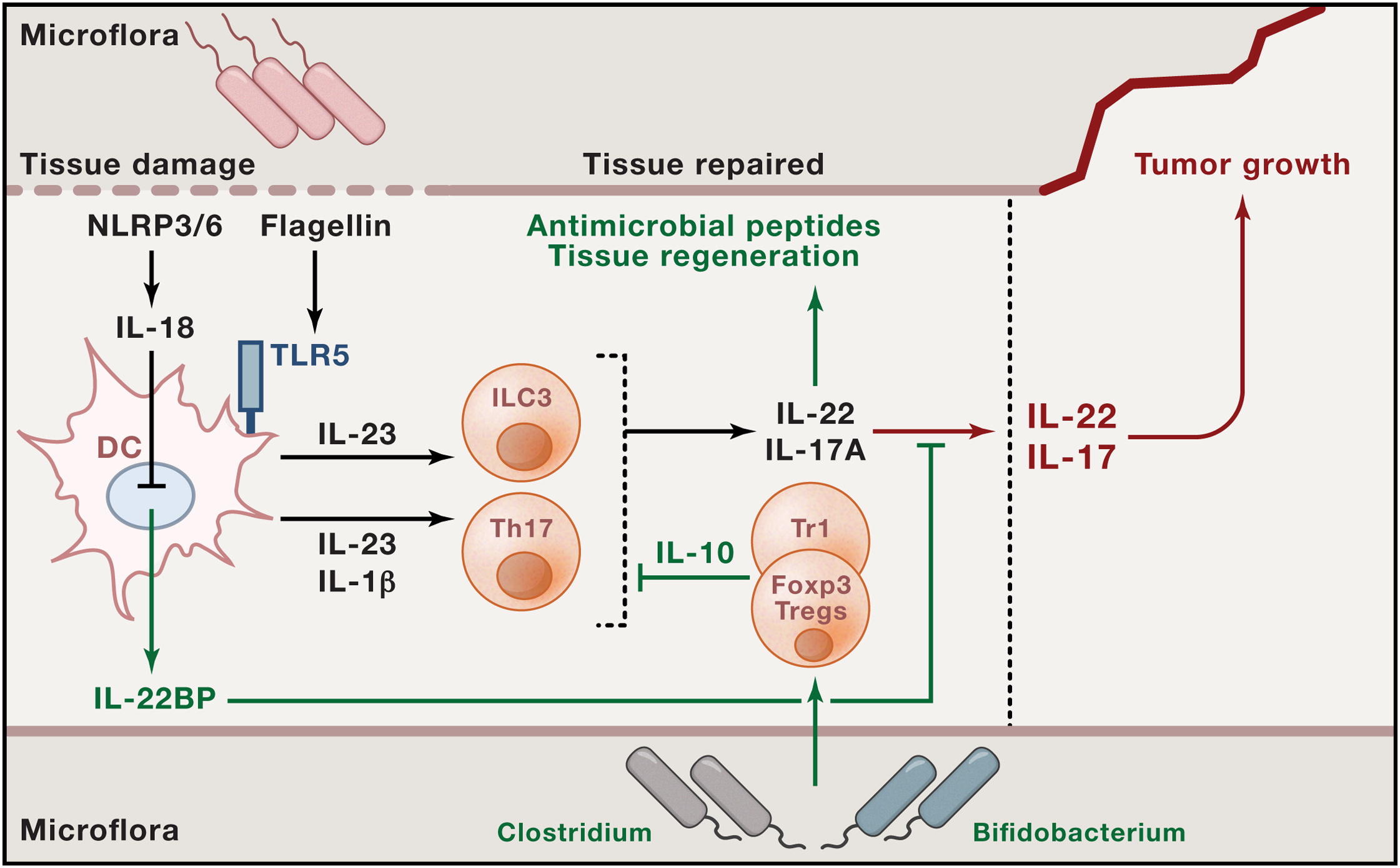
Fig1. Interplay between Microbes, Proinflammatory, and Anti-Inflammatory Immune Cells and Cytokines that May Influence Tumor Growth
Previous studies show that upon tissue damage and bacterial invasion, NLRP3/NLRP6 activates IL-18, which down regulates IL-22BP production by intestinal DC. Moreover, when DC sense bacterial components via TLR5, they produce IL-23. This cytokine acts on ILCs and, together with IL1β on Th17 cells, promotes the production of IL-17A and IL-22. Adequate levels of these two cytokine promote antimicrobial peptide secretion and tissue regeneration; their levels are kept in check by IL-22BP and IL-10 from regulatory T cells, which, in turn, may be induced by specific commensal bacteria. If IL-17A and IL-22 are not controlled, they can promote tumorigenesis. We could conclude accordingly that serious IBD may lead to tumor. Thus, it is necessary to focus on IBD healing. Inflammatory bowel diseases (IBD; Crohn’s disease (CD), ulcerative colitis(UC)) are chronic relapsing diseases that lead to structural damage with destruction of the bowel wall. Clinically, IBD patients can suffer from chronic diarrhea, malabsorption, weight loss, rectal bleeding, abdominal pain, stenoses, abscesses, and fistula formation, and many patients require surgery over time.
Fig2. Signaling events in inflammatory bowel diseases.
Although clinical IBD studies in the past have mainly focused on improvement of diarrhea and abdominal pain, mucosal healing on endoscopy has recently emerged as a key treatment goal in IBD. Mucosal healing in IBD could also result in effective disease control, more frequent steroid-free remission of disease, lower rates of hospitalization and surgery, and improved quality of live as compared with conventional treatment goals. In spite of the fact that not all groups agree on the relevance of mucosal healing in the absence of large prospective intervention studies, mucosal healing has become a key end point in clinical trials and predicts sustained clinical remission and resection-free survival of IBD patients. It was found that mucosal healing relies on the coordinated activity of intestinal epithelial cells (IECs), goblet cells, and Paneth cells for improvement of intestinal barrier function. Epithelial restitution is followed by additional steps in wound healing such as increased epithelial cell proliferation and differentiation. For example, IL-6 and IL-22 are known to boost proliferation of IECs by activation of the transcription factor STAT3. In contrast, the proinflammatory cytokines IFN-g and tumor necrosis factor (TNF) were found to block IEC proliferation and impaired wound healing by favoring IEC apoptosis. TLRs are additional key regulator of wound healing. In the healthy mucosa, TLRs act as innate receptors for pathogenassociated molecular patterns in commensal bacteria. Classical drugs for IBD are focus on the immune system. For instance, corticosteroids, cyclosporin A and tacrolimus are known to suppress proinflammatory cytokine production by T cells. NFATc2 controls IL-6 production and T-cell survival in colitis. Azathioprine and 6-mercaptopurine are effective in maintenance of remission in both CD and UC by blocking T-cell activation. As not all IBD patients respond to this type of therapy and as these drugs may cause severe side effects in some patients, it is of high clinical relevance to define novel drugs that block specific signaling pathways in cells of the adaptive immune system. Anti-TNF antibody therapy has been shown to improve IEC-controlled barrier function in IBD patients, suggesting that this treatment may have a profound effect on IEC function in vivo.The clinical development of neutralizing antibodies against TNF has been a crucial milestone for IBD therapy. Although several potential mechanisms of action have been proposed for anti-TNF therapy in IBD (e.g. barrier improvement, direct cytotoxicity, induction of regulatory macrophages, and T cells), several lines of evidence suggest that the induction of T-cell apoptosis is a key mechanism of action of anti-TNF antibodies. The clinically effective anti-TNF antibodies adalimumab, certolizumab pegol, and infliximab mainly work by blocking mTNF signaling to induce T-cell death. Thus, failure to anti-TNF therapy may occur in patients with low amounts of mTNF þ lamina propria cells due to TNF-independent gut inflammation. The findings that anti-TNF therapy is not effective in a relevant subgroup of IBD patients have led to an intensive search for novel therapeutic approaches.Anti-IFN-g therapy with the humanized antibody fontolizumab could decrease CRP levels, but could not result in major improvement of CD symptoms in moderate-tosevere disease activity.Another interesting cytokine for IBD therapy consists of IL-6. An initial study using the humanized anti-IL-6R antibody MRA showed a clinical response in 80% of the CD patients given biweekly MRA as compared with 31% in the placebo group. However, no effects on mucosal healing were noted after 12 weeks.In addition to targeting of IFN-g and IL-17A, blockade of the CD28/B7.1-2 interactions with the monoclonal antibody abatacept was not successful for CD and UC therapy. Furthermore, the anti-CD3 antibody visilizumab was not effective for the treatment of corticosteroid-refractory UC. Given these disappointing findings, new concepts for suppression of proinflammatory cytokines and T-cell activation are needed.There was research shows that local administration of IL-10 could be an alternative as well as a treatment with bacterial strains that induce intestinal IL-10 production, but these approaches require further development. Additional interesting anti-inflammatory cytokines for IBD therapy are IL-22, IL-35, and TGF-b. IL-22 induces STAT3 activation in IECs and favors IEC proliferation and colonic wound healing in vivo. Treatment with recombinant TGF-b has not been considered for IBD therapy as yet, mainly because of concerns regarding the well-known fibrogenic and cancer-supporting effects of this cytokine. One recent study found out how the therapies promote mucosal healing through inflammatory responding and identify the mechanism that links inflammation and barrier dysfunction. The proinflammatory cytokine tumor necrosis factor–a (TNF-a) is the primary target of IBD therapy at present. However, what determines the response to anti–TNF-a therapy in IBD remains unclear. Interleukin-22 (IL-22) is up-regulated in the intestine in patients with IBD to promote mucosal healing. However, uncontrolled IL-22 would lead to intestinal pathogenesis, even tumor. IL-22 binding protein (IL-22BP) exerts this control by specifically binding IL-22 and preventing it from binding membrane-bound IL-22 receptor 1 (IL-22R1). Regulation of IL-22 and IL-22BP controls homeostasis in the intestine; however, the role of IL-22BP in humans in IBD is uncertain. The mRNA expression for the inflammatory markers IL-22, IL-17A, and interferon-g (IFN-g) increased in the colon and terminal ileum of patients with active CD and UC. The IL22BP mRNA was also increased in the colon of these patients with less expansion. The study also observed dendritic cells (DCs), eosinophils and CD4+CD3+CD11c– T cells in the small intestine express high levels of IL-22BP. Il22bp–/+ mice transferred Il22bp-deficient T cells were largely protected from colitis showed that T cell–derived IL-22BP plays an important pathogenic role in multiple mouse IBD models. Rag1−/−Il22−/− mice transferred with Il22−/− and Il22−/−Il22bp−/− CD4+CD25–CD45RBhigh T cells caused equally severe colitis showed that IL-22BP aggravates colitis by blocking IL-22.
Fig3. A pathogenic role of CD4+ Tcell–derived IL-22BP in a murine colitis model.
The author found a positive correlation between IL22BP and TNFa expression in the intestine of IBD patients with active disease; however, genes encoding other cytokines, such as IL18, IL6, and IL23, did not correlate with IL22BP expression. Although IL22 also positively correlated with IL22BP, IL-22 does not regulate IL-22BP directly. Accordingly, the author wanted to find the link between IL-22BP and anti-TNF-therapy. IL22BP expression was markedly reduced in CD4+ T cells of IBD patients who were responsive to anti–TNF-a treatment, compared with those of patients on other medications. And other T cell signature cytokines and transcriptional regulators have no significant changes. However, IL22bp expression is not significantly reduced in Tnfr1- and Tnfr2-deficient T cells in the transfer colitis model and TNF-a did not induce IL22BP in T cells in vitro. Taken together, these data suggest that TNF-a might regulate IL-22BP in an indirect manner that is as yet unknown. Il22+/− mice transferred with wild-type and Il22−/− CD4+CD25–CD45RBhigh T cells respectively, and treated the mice with anti–TNF-a upon colitis development. Anti–TNF-a treatment was not effective in the Il22–deficient environment, but anti–TNF-a therapy significantly reduced colitis severity in the Il22–sufficient environment. Further the author treated the mice with anti–TNF-a upon colitis development in IL-22BP deficient environment, these mice developed a mild colitis, which, however, was not further improved by anti–TNF-a therapy. In conclusion, these data indicate that one mechanism whereby anti–TNF-a therapy may reduce disease activity is by down-regulating expression of IL-22BP; thus allowing IL-22–induced mucosal healing. So targeting IL-22BP directly might allow for a more effective and specific therapy for IBD without invoking the undesirable and potentially dangerous side effects of anti–TNF-a. Additionally, as mentioned before, uncontrolled IL-22 may increase patients’ risk of developing cancer. Therefore, long-term anti–IL-22BP treatment might have similar effects, which the doctors should pay attention to. Reference: 1. The Fire Within: Microbes Inflame Tumors. N. Gagliani, B. Hu, S. Huber, E. Elinav, R. A. Flavell, Cell 157, 776–783 (2014). 2. New targets for mucosal healing and therapy in inflammatory bowel diseases. M. F. Neurath, Mucosal Immunol. 7, 6–19 (2014). 3. A pathogenic role for T cell–derived IL-22BP in inflammatory bowel disease. Penelope Pelczar, Mario Witkowski, Laura Garcia Perez, et al. Science 354, 6310, 358-362 (2016)