Uncategorized Thursday, 2025/11/13
The team is now testing whether the insights gained from their study of FOXP2 can assist in treating Huntington's disease and other polyglutamine diseases. Ultimately, they aim to design drugs that can mimic the anti-aggregation effects of DNA binding and phosphorylation.
In deadly genetic disorders like Huntington's disease and spinocerebellar ataxias, proteins form long chains of repeat sequences that stick together like Velcro.
When these proteins aggregate in the brain, they damage and kill neurons, resulting in severe cognitive decline. Clinicians currently have no therapies targeting these pathological protein aggregates, and there is no cure for Huntington's disease and related disorders. However, a mystery persists: some proteins naturally have the same or even longer repeat regions (known as polyglutamine expansions) but do not aggregate at all.
A study published in the journal Cell by Stanford Medicine delved into this mystery. The research team focused on FOXP2 , a well-known gene closely linked to human speech and language function. Surprisingly, the protein contains one of the longest known polyglutamine extension sequences in the human body, yet it usually does not aggregate or cause disease.
Featured Proteins
| Cat.No. # | Product Name | Source (Host) | Species | Tag | Protein Length | Price |
|---|---|---|---|---|---|---|
| FOXP2-12985H | Recombinant Human FOXP2, His-tagged | E.coli | Human | His | 1-56a.a. | |
| FOXP2-1745R | Recombinant Rhesus monkey FOXP2 Protein, His-tagged | Mammalian Cells | Rhesus macaque | His |
|
|
| FOXP2-2389R | Recombinant Rat FOXP2 Protein | Mammalian Cells | Rat | His |
|
|
| FOXP2-3220Z | Recombinant Zebrafish FOXP2 | Mammalian Cells | Zebrafish | His |
|
|
| FOXP2-6011M | Recombinant Mouse FOXP2 Protein | Mammalian Cells | Mouse | His |
|
|
| FOXP2-093H | Recombinant Human forkhead box P2 Protein, His tagged | E.coli | Human | His | 478-596aa |
|
| FOXP2-1523H | Recombinant Human FOXP2 Protein, His&GST-tagged | E.coli | Human | GST&His | Met1-Ile244 |
|
| FOXP2-1567R | Recombinant Rhesus Macaque FOXP2 Protein, His (Fc)-Avi-tagged | HEK293 | Rhesus macaque | Avi&Fc&His |
|
|
| FOXP2-1866HFL | Recombinant Full Length Human FOXP2 Protein, C-Flag-tagged | Mammalian Cells | Human | Flag | Full L. |
|
"We're very excited to figure out why this is," said Dr. Joanna Wysocka, the Lorry Lokey Professor and professor of chemical and systems biology & developmental biology. "Could this protein teach us how to stop protein aggregation in Huntington's disease? This was a seemingly niche basic science problem that turns out to have significant implications for disease treatment."
What makes FOXP2 special?
FOXP2 is often called the "language gene." Mutations in FOXP2 are associated with a disorder known as apraxia of speech, where individuals can understand language but struggle to coordinate the movements of lips, tongue, and jaw necessary for speaking. Scientists speculate that changes in FOXP2 may explain how humans initially evolved the ability to speak.
FOXP2 is nearly identical in most mammals, but there are two minute features unique to humans. When mice possess the human version of the gene, they can produce more complex sounds and form connections in their brains more readily.
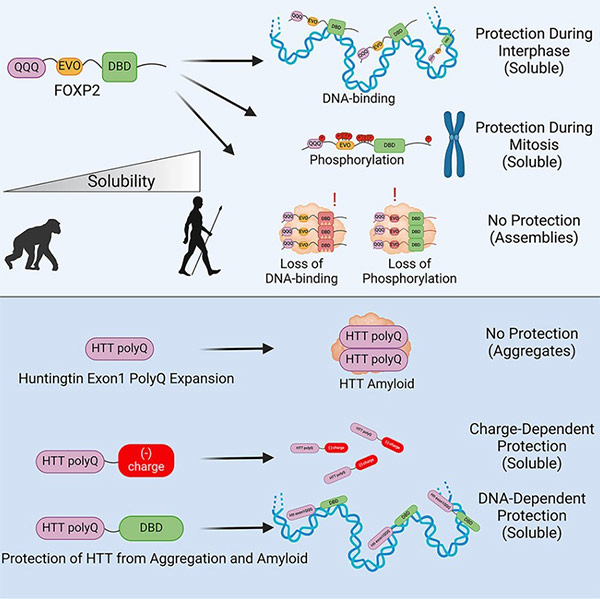
Fig1. Graphical abstract
Aggregation association
Huntington's disease is one of nine known disorders caused by long chains of polyglutamine extensions, essentially DNA sequences of three repetitive letters. When the polyglutamine repeat count in the Huntington gene surpasses 36, it typically results in disease. Symptoms usually manifest in middle adulthood and include involuntary movements, memory issues, and mood changes. The disorder is progressive and always fatal.
Surprisingly, FOXP2 inherently has a polyglutamine extension that repeats consecutively over 40 times, along with another 10 nearby repeats – yet it does not aggregate. Stanford Medicine's team aimed to understand why.
"We know FOXP2 is active in neurons due to its role in language," Wysocka explained. "Why can the brain tolerate such a long polyglutamine extension in FOXP2 but not shorter ones in Huntington's disease?"
This question deeply intrigued Dr. Shady Saad, a postdoctoral researcher co-mentored by Wysocka and Dr. Daniel Jarosz, an expert on protein aggregation and associate professor of chemical and systems biology & developmental biology.
"This protein sequence is so extreme that it seemed to require multiple mechanisms to prevent its aggregation right from the start," said Jarosz.
The protective factors of FOXP2
Through a series of laboratory experiments, Saad identified two primary reasons why FOXP2 typically does not aggregate. First , it can bind to DNA. FOXP2 is a DNA-binding protein that regulates genes. In most cells, FOXP2 is bound to DNA, which seems to help disperse it – preventing clumping and self-adherence. When the protein residues responsible for FOXP2's DNA binding are disrupted (as seen in apraxia of speech manifestations), the protein begins to aggregate.
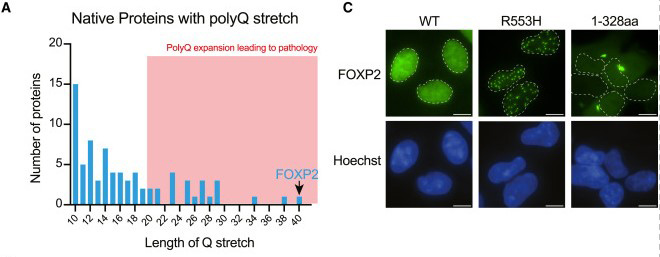
Fig2. FOXP2 depends on DNA binding for solubility. (A) PolyQ tract length distribution in the human proteome. Lengths associated with neurodegenerative disease are shaded in red. (C) Representative fluorescence micrographs of HEK293 cells with stable expression of wild-type FOXP2 and two dyspraxia-associated mutants (R553H and R328X) fused to EGFP. Contours of nuclei denoted with white dashed lines.
"We believe DNA acts as a rigid polymer, with other possible attached factors, helping disperse FOXP2, reducing the chances of individual proteins coalescing and forming aggregates," Wysocka explained.
Second , during cell division, FOXP2 gains a charged chemical "coat." As cells begin to divide, most transcription factors detach from DNA. So why doesn't FOXP2 start sticking together at that time?
As it turns out, during this phase, the protein gains another form of anti-aggregation protection: structurally, phosphate chemical groups are added to it. These negatively charged chemical coatings prevent FOXP2 protein molecules from sticking to one another. When FOXP2's ability to undergo phosphorylation is blocked, the protein starts to aggregate like the disease-associated proteins.
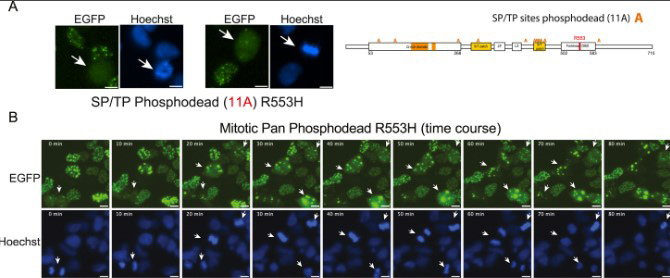
Fig3. Concerted function of FOXP2 phosphorylation sites in mitotic solubility, (A) Left: fluorescence micrographs of mitotic cells (white arrows) stably expressing FOXP2 R553H SP/TP 11A, showing complete (left) or partial (right) assembly dissolution. Right: schematic of the 11A phosphodead mutant in which SP/TP are mutated to alanine in the R553H background. (B) Representative time-lapse images of asynchronous culture stably expressing FOXP2 R553H pan-phosphodead mutant. Assemblies of this mutant do not solubilize in mitotic cells (white arrows).
Does this help in treating diseases?
The combined effects of DNA binding and phosphorylation function as a natural anti-aggregation system for FOXP2. The researchers wanted to know if they could use the same strategy against pathogenic proteins associated with polyglutamine expansion diseases. "Because FOXP2 and Huntington's proteins share the same aggregating characteristics, we wondered if our anti-aggregation strategies could dissolve pathological proteins," Saad explained.
To test this idea, the team fused the easily aggregating Huntington's protein with the protective features of FOXP2. Surprisingly, these additives were sufficient to reduce – and in some cases completely dissolve – toxic aggregates. Their findings included:
(1) adding a DNA-binding tag to the Huntington protein helps break them down within cells;
(2) mimicking the negatively charged phosphate molecules on FOXP2 also worked in preventing sticky aggregates from forming;
(3) even the stubborn, insoluble aggregates known as amyloids formed in the brains of Huntington's disease patients became more dissolvable after adding these traits.
"I was very surprised at how well it worked," Wysocka said. "These are notoriously difficult aggregates to break apart. The fact that they started dissolving is truly exciting."
"Finding out that our cells naturally have the capacity to dissolve these aggregates was quite unexpected," Jarosz said. "It's been hiding in plain sight all along."
"The results were black-and-white; the polyglutamine amyloids disappeared, and that was a happy moment," Saad noted.
Evolutionary insights
When the team compared FOXP2 proteins from humans, chimpanzees, and mice, the story came full circle. They found the human version of FOXP2 significantly more soluble – less prone to aggregating – compared to versions from other species.
It turns out, those same changes in FOXP2 that enhance vocal abilities and brain plasticity in mice also make the protein less likely to aggregate. In other words, the evolutionary adjustments that helped humans develop language capabilities might have also rendered FOXP2 safer when present at high levels in the brain.
"This raises the tantalizing possibility that increased solubility might have allowed our brains to boost FOXP2 levels without causing harm," Wysocka said. "This may have paved the way for the evolution of human speech."
What's next for FOXP2 research?
The team is now testing if the insights gained from FOXP2 research can help treat Huntington's disease and other polyglutamine diseases. Ultimately, they hope to design drugs that can mimic the anti-aggregation effects of DNA binding and phosphorylation.
"We've developed a whole new approach to targeting these diseases by addressing their root causes," Saad shared. "I'm thrilled by the potential outcomes."
"We initially started exploring a basic science question about evolution," Wysocka stated. "Now, we're thinking about how to treat an incurable disease. That's the power of foundational research – it can lead you to places you never anticipated."
Related Products & Services
- Neurodegenerative Disease
- Adhesion Molecules
- Blood-brain Barrier Permeability
- Neurotrophic Factors and Receptors
- Protein Engineering Services
- Protein Interaction Service
- Protein Expression and Purification Services
- Drug Discovery Screening
- Protein Pathway Profiling
Reference
- Shady Saad et al, DNA binding and mitotic phosphorylation protect polyglutamine proteins from assembly formation, Cell (2025). DOI: 10.1016/j.cell.2025.03.031.