Uncategorized Tuesday, 2025/11/25
Researchers have developed an ingenious method to successfully “turn back time” and glimpse the true state of cells that are destined to be infected before they “get into trouble”.
Imagine a crowded room where someone sneezes and the cold virus instantly fills the air. A few days later, some people are fine, while others start to sniffle and cough. We usually attribute this to differences in individual immunity. But if we switch the scene to the microscopic world and enter a tissue composed of tens of thousands of almost identical cells, a more fundamental and puzzling question arises: When the virus army is at the gate, why are some cells infected while their genetically identical “neighbors” next to them are spared?
This is not a coincidence. For a long time, researchers have observed that even in the most homogeneous cell culture systems, viral infections show significant heterogeneity. What kind of law of life lies behind this phenomenon? Is it the random collision of the virus, or has the cell itself already been “predestined”?
Answering this question is far more difficult than imagined. The biggest obstacle is that once the virus enters the cell, it will quickly “tamper” with the host's life program, shutting down its normal transcription and translation, and turning it into a factory for viral replication. When we try to find the reason for its susceptibility by analyzing the gene expression of infected cells, what we see is already the state “remodeled” by the virus, not its “bare face” before infection. It's like a murder case, when we arrive at the scene, all clues have been destroyed by the murderer, and we can't trace the original truth.
However, the research report in “Cell” titled “Single-cell susceptibility to viral infection is driven by variable cell states” provided an unprecedented key to solving this puzzle. Researchers developed an ingenious method to successfully “turn back time” and glimpse the true state of cells that are destined to be infected before they “get into trouble”. Their findings not only profoundly reveal the intrinsic logic of viral infections but also point the way for us to understand individual differences in diseases and develop new antiviral strategies.
Pressing the "Rewind" Button for Cell Fate: An Ingenious Tracing Technology
To crack the “chicken or egg” dilemma, whether the cell state leads to infection or infection changes the cell state, the key is to have a “prophet” that can see through the intrinsic characteristics of cells before infection occurs. Traditional methods can't do this, but the research team took a different path and used a single-cell clone tracing technology called “Rewind” to achieve “time-space travel” for cell fate.
The essence of this technology can be understood as follows:
First, researchers constructed a huge “barcode library”. These DNA barcodes were randomly integrated into the genomes of single cells through lentivirus vectors. Each cell and its offspring (i.e., a clone) would have a unique, heritable “ID card”.
Next, they let these cells with “ID cards” proliferate. When a cell divides into two, they become a pair of “identical twins” with the same genetic background and “ID card”. The researchers cleverly split these cell “twins” into two groups:
One group (archive group): Immediately perform single-cell RNA sequencing (scRNA-seq). This is equivalent to taking a detailed “molecular snapshot” of each cell clone at a certain time point, recording its current gene expression state, that is, its “cell state”.
The other group (experimental group): Expose to the environment of SARS-CoV-2 . After a period of time, use the method of fluorescently labeling viral RNA to separate infected and uninfected cells.
Finally, and most importantly, researchers extracted DNA from the “infected” and “uninfected” cell populations in the experimental group and read their “ID card” barcodes. By matching these barcodes with the “molecular snapshots” of the archive group, they could precisely answer the core question: What were the “twin brothers” of the cells that were eventually infected by the virus like before infection?
This method is like having a cell fate video recorder. It first records the initial state of each cell, then fast-forwards to show their process of experiencing viral infection, and finally “rewinds” to carefully examine the original footage of the “infected” cells. In this way, researchers successfully bypassed the virus's interference with cells and directly hit the intrinsic essence that determines susceptibility.
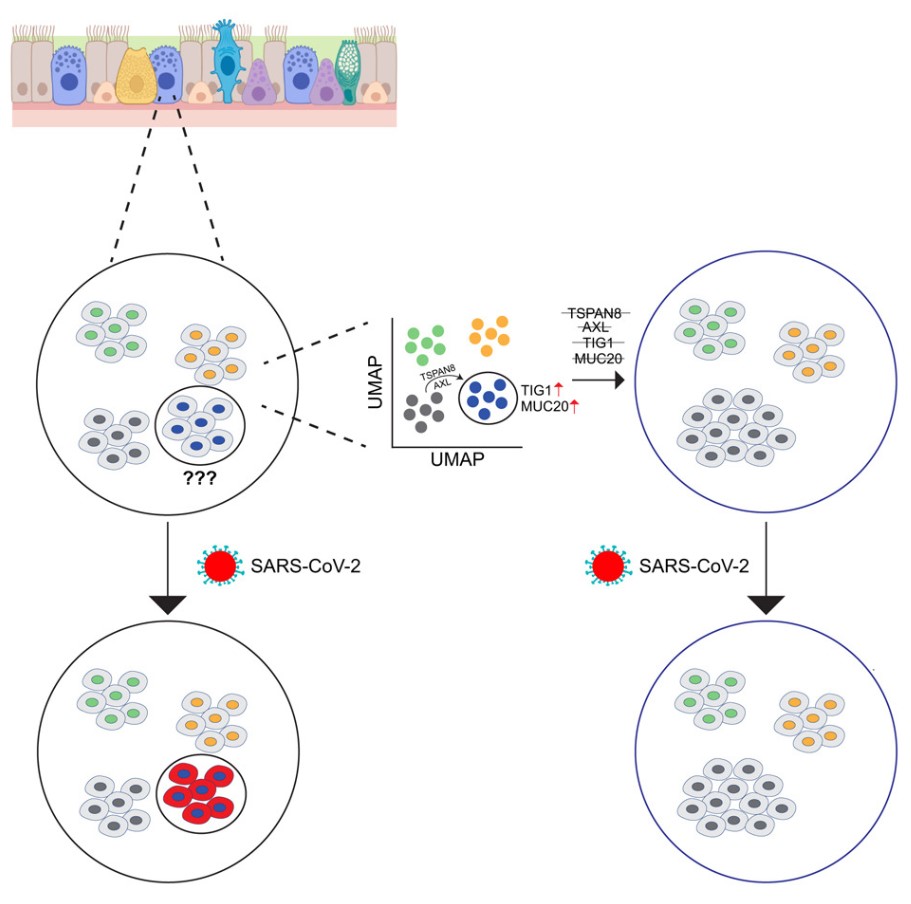
The Portrait of the "Susceptible": The Emergence of the High Expression State of TIG1
Using this powerful “Rewind” system, researchers used the human lung adenocarcinoma cell line Calu-3 as a model. This is a classic model for studying respiratory viruses because it can well simulate the characteristics of human respiratory epithelial cells. They found that 24 and 48 hours after infection with SARS-CoV-2, the “ID cards” of the infected cells were not randomly distributed in the cell atlas of the archive group, but were significantly enriched in two specific cell subgroups.
One subgroup is rapidly proliferating cells, which is in line with our common understanding that active cells may be more easily exploited by viruses. But more interestingly, the other subgroup is marked by the high expression of a gene called TIG1 (Tazarotene-induced gene 1) . This cell subgroup, which researchers call the “TIG1-high” state, seems to be the “ideal prey” of SARS-CoV-2.
Further analysis revealed that “TIG1-high” is not just the elevation of a single gene, but the coordinated activation of an entire gene expression program, which is closely related to the retinoic acid signaling pathway. In sharp contrast, another cell subgroup that expresses a large number of interferon-stimulated genes (Interferon-stimulated genes, ISGs), that is, cells in the “JAK/STAT” signaling activation state, show natural resistance, with almost no infected cell clones coming from this subgroup. This indicates that the intrinsic, spontaneous gene expression patterns of cells have already predetermined their fate in front of the virus: some cells have turned on the “welcome mode” (such as TIG1-high), while others have activated the “defense mode” (such as JAK/STAT activation).
To verify whether these genes are not just “bystander” markers but “masterminds” that determine fate, researchers conducted a series of gene knockout experiments. They used CRISPR-Cas9 technology to systematically knock out several genes highly expressed in the “TIG1-high” state, including AXL , TSPAN8 , MUC20 , and TIG1 itself. The results were very clear: after knocking out these genes, the infection rate of cells with SARS-CoV-2 all significantly decreased. This strongly proves that these genes in the “TIG1-high” state jointly build a “microenvironment” favorable for viral invasion and replication.
Related Proteins
Beyond ACE2: Viral Receptors Are Not the Only "Passport" for Susceptibility
When it comes to SARS-CoV-2 infection, almost everyone thinks of its “key” - the ACE2 receptor (Angiotensin-converting enzyme 2). Theoretically, the more ACE2 protein on the cell surface, the greater the chance for the virus to enter the cell. So, is the difference in cell susceptibility simply due to differences in ACE2 expression levels?
Researchers delved into this question and found that the story is far more complex.
Through single-cell sequencing and RNA in situ hybridization (smFISH) techniques, they found that in the Calu-3 cell population, cells that could detect ACE2 mRNA were very rare. Even when they turned to detecting ACE2 protein, they found that although about 9% of cells expressed high levels of ACE2 protein, the proportion of actually infected cells was much lower (for example, only 1.5% after 24 hours). This indicates that high expression of ACE2 is a necessary condition for cell infection, but far from a sufficient condition.
Who are the real “chosen ones”, the most susceptible cells?
Researchers used immunofluorescence and smFISH in combination to simultaneously detect ACE2 protein and TIG1 mRNA in single cells. The data revealed a surprising phenomenon: There is a very strong positive correlation between the high expression of ACE2 protein and the high expression of TIG1. The probability of a cell being both ACE2-high and TIG1-high is 33.16 times that of a random situation.
However, even so, only about 28.7% of ACE2-high cells are also TIG1-high. This means that among all cells holding the “entry ticket” (ACE2-high), only a small part simultaneously activates the “VIP channel” of “TIG1-high”, and they are the virus's top targets. In comparison, another mucin gene, MUC5AC , although also has a certain correlation with ACE2 expression, its association strength (probability of 2.63 times) is much lower than that of TIG1.
This finding overturns the simple model that previously attributed viral susceptibility mainly to receptor expression levels. It reveals a more refined regulatory level: Viral susceptibility is determined by a multi-gene, multi-dimensional “cell state”, not just the presence or absence of a single receptor molecule. ACE2 may only open the first door, while the “TIG1-high” state paves the way for the virus's subsequent invasion and replication.
Related Proteins
| Cat.No. # | Product Name | Source (Host) | Species | Tag | Protein Length | Price |
|---|---|---|---|---|---|---|
| ACE2-736H |
Active Recombinant Human ACE2 protein, His-tagged


|
HEK293 | Human | His | 18-740 aa | |
| ACE2-68H |
Active Recombinant Human ACE2, Fc-tagged
|
HEK293 | Human | Fc | 18-740 aa | |
| ACE2-736HA |
Active Recombinant Human ACE2, His-tagged, APC labeled
|
HEK293 | Human | His | ||
| Ace2-1183M |
Recombinant Mouse Ace2 protein, His&hFc-tagged
|
HEK293 | Mouse | Fc&His | Met1-Thr740 | |
| ACE2-185H |
Active Recombinant Human ACE2 protein, mFc-tagged
|
HEK293 | Human | mFc | Gln 18 - Ser 740 | |
| Ace2-1184M | Recombinant Mouse ACE2 protein(Met1-Thr740), His-tagged | HEK293 | Mouse | His | 1-740 a.a. | |
| MUC5AC-17H | Recombinant Human MUC5AC Protein, His-tagged | E.coli | Human | His | Tyr5568~His5654 | |
| MUC5AC-16H | Recombinant Human MUC5AC protein, His-tagged | E.coli | Human | His |
|
|
| MUC5AC-28542TH | Recombinant Human MUC5AC protein, GST-tagged | Wheat Germ | Human | GST | 351-450 a.a. |
|
Unveiling the Regulatory Network of the “Susceptible State”: Who is the Puppet Master?
Since the “TIG1-high” state is a complex state composed of multiple genes, is there any regulatory relationship among these genes? Are they “a scattered group” or an organized “crime syndicate”?
To explore this question, researchers applied computational methods of pseudotime analysis. This method can infer the “trajectory” and “timeline” of cell state evolution based on the progressive changes in single-cell gene expression profiles. The analysis results showed that there is a clear path from ordinary proliferating cells to the susceptible “TIG1-high” state.
Along this path, the expression of different genes shows distinct temporality: The expression of genes AXL and TSPAN8 increases early in the trajectory, followed by TIG1, and genes such as CEACAM1 , MUC20, and CTSS reach their peaks at the end of the trajectory. This temporality suggests a potential regulatory cascade: Early genes (such as AXL, TSPAN8) may act as “masterminds” to initiate and regulate the expression of downstream genes (such as TIG1, MUC20), thereby collectively shaping the entire “susceptible state”.
This is not just a computational speculation. Researchers once again verified this hypothesis through gene knockout experiments. They found that:
Knocking out the upstream AXL or TSPAN8 not only eliminated their own expression but also significantly reduced the expression levels of downstream TIG1 and MUC20. For example, after knocking out AXL, the proportion of cells with high TIG1 expression dropped sharply from 9.9% to 1.0%, and the proportion of cells with high MUC20 expression also decreased from 10.0% to 2.0%.
Knocking out the intermediate TIG1 led to a decrease in downstream MUC20 expression, but did not affect the more upstream AXL or TSPAN8.
Knocking out the most downstream CEACAM1 or CTSS had almost no effect on the expression of any other “susceptible genes”.
This series of interlocking evidence clearly outlines a regulatory network. AXL and TSPAN8 are like two “switches”; once pressed, they initiate a programmed process leading to the “TIG1-high” state. This finding is of great significance because it suggests that we might be able to dismantle the entire “susceptible state” by intervening in these upstream “mastermind” genes, thereby achieving broad-spectrum prevention of viral infections, rather than just targeting a downstream executor.
Related Proteins
| Cat.No. # | Product Name | Source (Host) | Species | Tag | Protein Length | Price |
|---|---|---|---|---|---|---|
| CEACAM1-11079H | Recombinant Human CEACAM1, His-tagged | E.coli | Human | His | 317-459a.a. | |
| Ceacam1-3312M | Recombinant Mouse Ceacam1 Protein, His tagged | HEK293 | Mouse | His | Met 1-Gly 428 | |
| CEACAM1-662H | Active Recombinant Human CEACAM1 protein, His-tagged | HEK293 | Human | His | 35-428 aa | |
| CEACAM1-663H |
Active Recombinant Human CEACAM1 protein, His&hFc-tagged
|
HEK293 | Human | Fc&His | 1-428 aa | |
| Ceacam1-3312MAF488 | Recombinant Mouse Ceacam1 Protein, His-tagged, Alexa Fluor 488 conjugated | HEK293 | Mouse | His | 405 | |
| Ceacam1-3312MAF555 | Recombinant Mouse Ceacam1 Protein, His-tagged, Alexa Fluor 555 conjugated | HEK293 | Mouse | His | 405 | |
| CTSS-911M |
Active Recombinant Mouse CTSS Protein, His-tagged
|
HEK293 | Mouse | His | Full L. 1-340 a.a. |
|
| Ctss-1113R | Recombinant Rat Ctss protein(Met1-Ile318), His-tagged | HEK293 | Rat | His | 1-318 a.a. |
|
| CTSS-1651H | Recombinant Human Cathepsin S | Human Spleen | Human | Non |
|
From Petri Dish to Real World: Do These “Susceptible Cells” Exist in the Human Body?
Is the “TIG1-high” susceptible state found in Calu-3 cells merely a “special case” in the petri dish, or does it also play a key role in real human lungs?
To answer this question, researchers analyzed publicly available human lung single-cell sequencing databases. They first identified various epithelial cell types in the complex lung tissue, including basal cells, alveolar type I and II cells (AT1/AT2 cells), club cells, and ciliated cells.
The results were encouraging: The “TIG1-high” gene signature was indeed present in the human lung and was mainly enriched in club and ciliated cells. These two cell types are also the main targets of SARS-CoV-2 in the lung. This indicates a high degree of consistency between laboratory findings and real-world situations.
Further analysis revealed the potential link between this “susceptible state” and clinical diseases. Researchers compared lung tissue samples from healthy individuals and patients with idiopathic pulmonary fibrosis (IPF). IPF is a severe, chronic lung disease of unknown cause, and patients with IPF often have a worse prognosis after COVID-19 infection. The analysis found that in the lungs of IPF patients, not only was the proportion of ciliated cells increased, but the frequency of club and ciliated cells expressing high levels of TIG1 and MUC20 was also significantly higher than in healthy individuals.
Additionally, they found that in healthy individuals with a smoking history, the proportion of lung cells expressing high levels of TIG1 and MUC20 was also significantly higher than in non-smokers.
These findings serve as a bridge connecting the microscopic “cell state” with macroscopic clinical risk factors (such as underlying lung diseases and smoking). They provide a new explanation: Why are certain groups of people more susceptible to SARS-CoV-2 infection or more likely to develop severe illness? One possible reason is that their lungs, due to the influence of diseases or environmental factors, already have more cells in the “TIG1-high” susceptible state, preparing a “fertile ground” for viral invasion and spread.
Virus's “Mating Standards”: Why Does Influenza Virus “Favor” Another Type of Cell?
Is the “TIG1-high” state a “universal key” for all respiratory viruses, or do different viruses have their unique “tastes” and “mating standards”?
To explore viral specificity, the research team turned to another common respiratory pathogen, Influenza A virus. They repeated “Rewind” experiments similar to those with SARS-CoV-2 to track the state of cells susceptible to influenza virus infection before infection.
The results brought another surprise. The barcodes of influenza virus-infected cell clones were not enriched in the “TIG1-high” cell subgroup. Instead, they were significantly concentrated in a completely different cell subgroup. This subgroup was characterized by high expression of keratin 8 and 18 (KRT8, KRT18) and SPP1 , presenting a “transitional stem cell” state.
This finding is highly enlightening. It indicates that even within the same cell population (Calu-3), there coexist “susceptible states” specific to different viruses. SARS-CoV-2 prefers cells in the “TIG1-high” state, while the influenza virus favors transitional cells in the “KRT8-high” state. It's like two different hunters entering the same forest but tracking different prey.
This viral specificity is also reflected in different SARS-CoV-2 variants. Researchers compared the infection preferences of the original strain (WA1) and the Omicron variant (BA.1) for cells. They found that although cells in the “TIG1-high” state showed higher susceptibility to both strains, this preference was more pronounced when infecting the original strain. This may partly explain why different variants have different pathogenicities. Viral evolution may also involve fine-tuning of its preference for the host cell's “susceptible state”.
Related Proteins
| Cat.No. # | Product Name | Source (Host) | Species | Tag | Protein Length | Price |
|---|---|---|---|---|---|---|
| KRT8-144H |
Recombinant Human Keratin 8
|
E.coli | Human | Non | Full L. | |
| KRT8-10433Z | Recombinant Zebrafish KRT8 | Mammalian Cells | Zebrafish | His |
|
|
| KRT8-145H | Recombinant Human KRT8, His-tagged | E.coli | Human | His | 1-483aa |
|
| KRT18-12H | Recombinant Human KRT18 protein, His-tagged | E.coli | Human | His |
|
|
| KRT18-13H | Recombinant Human KRT18 protein, T7/His-tagged | E.coli | Human | His&T7 | 1 a.a. - 100 a.a. |
|
| SPP1-664H | Recombinant Human SPP1 protein, His-tagged | Mammalian Cells | Human | His | 17-314 aa | |
| SPP1-2089H | Recombinant Human SPP1 Protein, His (Fc)-Avi-tagged | HEK293 | Human | Avi&Fc&His | ||
| SPP1-4136H | Recombinant Human SPP1 Protein, His (Fc)-Avi-tagged | HEK293 | Human | Avi&Fc&His |
Beyond “Whether Infected,” Moving to Deep Thinking on “Why Susceptible”
This study published in Cell , with its ingenious experimental design and detailed data, paints a new picture of the interaction between viruses and host cells. It tells us that whether a cell is infected by a virus is far from being determined by the mere presence of viral receptors on the cell surface. Beneath the genetic background, there exists a dynamic dimension defined by intrinsic gene expression programs - the “cell state." These ever-changing cell states form the “micro-battlefield” of viral infections, determining who is “susceptible” and who is “resistant."
The significance of this work is multifaceted:
First, it provides a new perspective for understanding viral tropism. Why do viruses only infect specific types of tissues and cells? The traditional answer is receptor distribution. Now we know that even among receptor-positive cell populations, the virus's targets are carefully selected; they aim for cell subgroups in specific “susceptible states."
Second, it offers a molecular basis for explaining individual differences in diseases. Why do people exposed to the same virus have vastly different disease severities? In addition to macro-level differences in the immune system, the baseline level of cellular “susceptible states” at the microscopic level may also be a key factor. A person with a higher proportion of “TIG1-high” cells in the lungs may provide more “beachheads” for the virus at the initial stage of infection.
Finally, it paves the way for developing new antiviral strategies. Traditional antiviral drugs mainly target the virus itself (e.g., inhibiting its replicase or protease), which can easily lead to viral resistance. This study suggests that we might adopt a reverse approach: instead of directly attacking the virus, we could regulate the host through drug interventions to shift cells from a “susceptible state” to a “resistant state." For example, developing drugs that inhibit upstream regulatory factors such as AXL or TSPAN8 could potentially shut down the entire “TIG1-high” program, thereby “keeping the virus at bay." The beauty of life lies in its complexity and precision. From the fate choices of individual cells to an individual's health and diseases, and even to the evolution of a global pandemic, all follow profound biological logic. This study is a brilliant decoding of this logic. It makes us realize that in the face of viral attacks, cells are not innocent “naive sweethearts." Their intrinsic states have already written their own scripts in the unseen. Reading these scripts is the hope for us to overcome diseases.
Related Products & Services
- SARS-CoV-2 Proteins and Their Target Proteins
- Infectious Diseases Targets
- Immune Checkpoint Proteins
- Protein Engineering Services
- Protein Interaction Service
- Protein Expression and Purification Services
- Drug Discovery Screening
- Protein Pathway Profiling
Reference
- Single-cell susceptibility to viral infection is driven by variable cell states. Reffsin, Sam et al. Cell, Volume 0, Issue 0