Uncategorized Saturday, 2025/08/23
This new study provides the first detailed revelation of the structure of this steroid hormone receptor complex.
Imagine a drug that could block cancer without side effects or risks. This future might be closer due to a new study led by Dr. Raj Kumar, the Chair of the Department of Pharmaceutical and Biomedical Sciences at the University of Toledo College of Pharmacy and Pharmaceutical Sciences. The findings are published in the journal Nature Communications.
The Kumar laboratory has been focused on studying steroid hormone receptors (SHRs), which are among the most frequently targeted proteins in cancer treatment. Steroid hormones, like estrogen and testosterone, become targets because the levels of SHRs in cancer cells are higher than normal, potentially promoting the occurrence of breast, prostate, and ovarian cancers. However, since these anti-hormone drugs also bind to SHRs on other cells in the body, like those in the uterus and bones, they often cause side effects and risks. Kumar stated, "While trying to cure breast cancer, the action of estrogen on uterine tissue is increased, which can lead to uterine cancer."
He said, "The 'holy grail' of SHR-based endocrine cancer therapy is to restrict the action of SHRs to specific organs and gene targets." To achieve this, a better understanding of SHRs' structures and how they interact with other proteins is necessary.
Structural proteomics reveals isomeric differences in progesterone response element (PRE) binding.
This new study provides the first detailed revelation of the structure of this steroid hormone receptor complex. Kumar stated, "When we understand the entire structure of SHRs and how the associated proteins form this complex, it becomes possible to design drugs that can specifically target certain tissues." Differences in SHR complexes explain why hormones—and potential drugs—act differently in various types of cells and tissues.
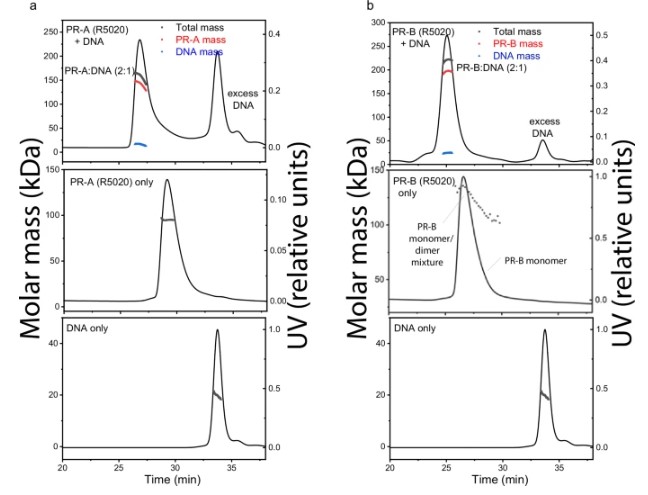
Fig1. DNA induces assembly of PR-A and PR-B.
A limited understanding of these structures has hindered the efforts to develop more effective, targeted drugs with minimal or no side effects. The Kumar team used novel proteomics techniques to reveal these structural differences.
Kumar stated, "To the best of our knowledge, this is the first time such detailed structural information has been shown in the structure of SHRs co-activator complexes. This study will significantly enhance our ability to predictively interfere with SHR signaling and paves the way for developing more effective drugs with minimal or no severe side effects for treating endocrine cancers."
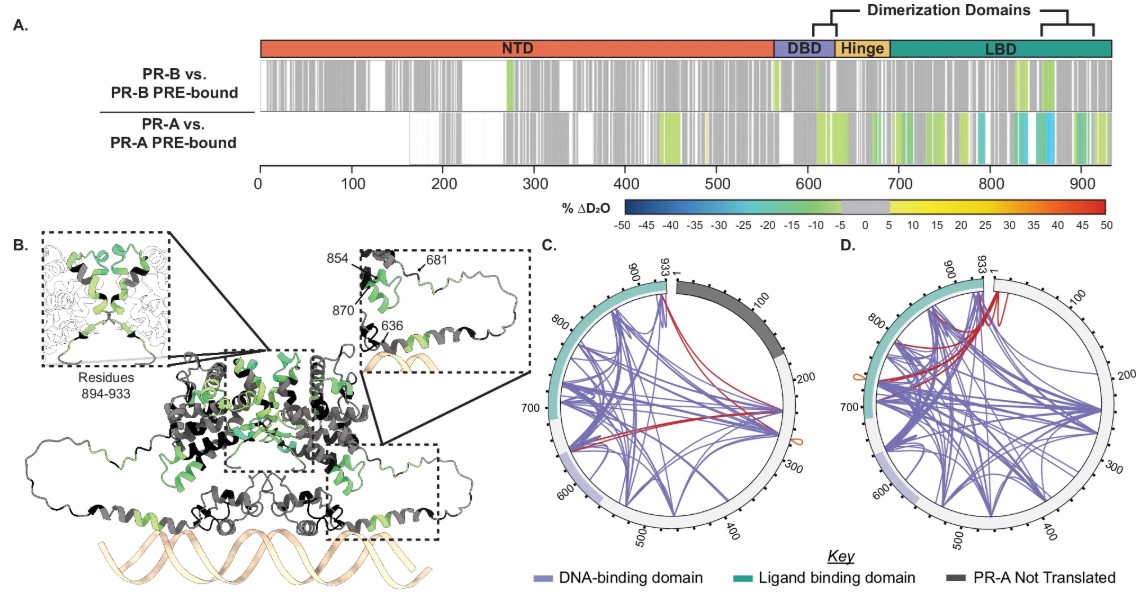
Fig2. Structural proteomics reveals isoform differences upon progesterone response element (PRE) binding.
Research Background
Progesterone Receptors (PR): Members of the nuclear receptor superfamily, these receptors regulate gene expression in the development of female reproductive tissues and in cancers such as breast and endometrial cancer.
PR-A vs. PR-B: PR-A is a truncated isoform (lacking the AF3 functional domain), whereas PR-B is a full-length isoform. The two have significantly different functions, though the structural mechanisms are unclear.
Technical Challenges: PR is a large, flexible protein, and traditional structural biology methods struggle to resolve its full-length dynamic interactions with co-activators (such as SRC3 and p300).
Research Highlights
- Structural Proteomics Strategy Technological Combination: By integrating Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS), Cross-Linking Mass Spectrometry (XL-MS), and AlphaFold3 modeling, this study for the first time resolves the dynamics of PR-coactivator-DNA complexes at amino acid resolution.
Sample Preparation: Full-length PR, SRC3, and p300 were expressed and purified at high purity in insect cells, ensuring native conformations and post-translational modifications (e.g., phosphorylation).
- Mechanism of PR-DNA Interaction DNA-induced Dimerization: Both PR-A and PR-B bind progesterone response elements (PRE) as dimers, but in the absence of DNA, PR-B partially exists as a dimer.
Conformational Changes: DNA binding transforms PR from a "compact" to an "extended" conformation, creating space for co-activator binding by separating the N-terminal domain (NTD) and ligand-binding domain (LBD).
- Sequential Recruitment of Co-activators "Pre-activation" by SRC3: PR-A preferentially binds to SRC3 (via NR-box 2) and can induce PR dimerization without DNA. PR-B interacts weakly with SRC3, relying on DNA binding to recruit SRC3.
"Stabilization" by p300: p300 further stabilizes the ternary complex (PR:SRC3:p300) by binding to SRC3's NR-box 3, forming a transcription activation platform. p300 can directly interact with PR's NTD and LBD independently of SRC3.
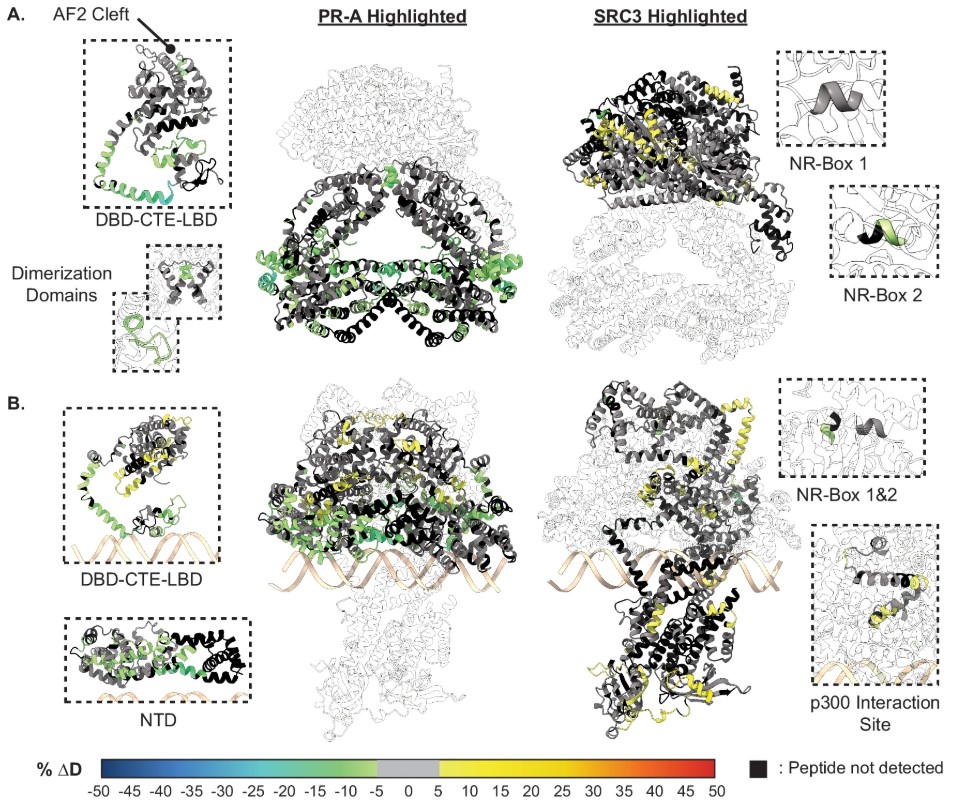
Fig3. SRC3 induces LBD changes to PR upon PRE addition.
- Unexpected Effects of Antagonist RU486 Complex Formation Maintained: PR-A/B bound to RU486 (a traditional antagonist) can still recruit SRC3 and p300, but interaction sites shift from the C-terminal (AF2) to the N-terminal, leading to transcription repression.
Mechanism Remodeled: Antagonists inhibit transcription by altering the spatial conformation of the complex (e.g., displacing the HAT domain from chromatin) rather than completely blocking co-activator binding.
- Functional Validation Mutation Experiments: Mutating SRC3's NR-box 2 (from LXXLL to LXXAA) significantly reduces PR transcriptional activity, underscoring its critical role.
Isoform Differences: PR-A has a stronger dependency on SRC3, possibly linked to poor prognosis associated with PR-A overexpression in breast cancer.
- Unexpected Effects of Antagonist RU486 Complex Formation Maintained: PR-A/B bound to RU486 (a traditional antagonist) can still recruit SRC3 and p300, but interaction sites shift from the C-terminal (AF2) to the N-terminal, leading to transcription repression.
Mechanism Remodeled: Antagonists inhibit transcription by altering the spatial conformation of the complex (e.g., displacing the HAT domain from chromatin) rather than completely blocking co-activator binding.
- Functional Validation Mutation Experiments: Mutating SRC3's NR-box 2 (from LXXLL to LXXAA) significantly reduces PR transcriptional activity, underscoring its critical role.
Isoform Differences: PR-A has a stronger dependency on SRC3, possibly linked to poor prognosis associated with PR-A overexpression in breast cancer.
Scientific Significance
Revealing Isoform-Specific Regulation of PR: PR-A and PR-B selectively bind co-activators through different conformations, explaining their tissue-specific functions.
Challenging the Classic Model: The antagonist RU486 does not simply "exclude" co-activators but inhibits transcription by remodeling complex conformation.
Therapeutic Implications: Targeting interaction sites between SRC3/p300 and PR, such as NR-box 2/3, may offer a new strategy for cancer therapy.
One-Sentence Summary
This study elucidates the sequential activation mechanism of progesterone receptor isoforms in coordination with DNA and co-activators through high-resolution structural proteomics, providing a key framework for understanding hormone receptor regulation and developing targeted therapeutics.
Related Products & Services
- Cytokines for Organoid Culture
- ADC Target Protein
- PROTAC Targets
- Cell and Gene Therapy
- Targets of CAR-T Cell Therapy
- Cancer Drug Targets
- Immune Checkpoint Proteins
- Protein Engineering Services
- Protein Interaction Service
- Protein Expression and Purification Services
- Drug Discovery Screening
- Protein Pathway Profiling
Reference
- Matthew D. Mann et al, Structural proteomics defines a sequential priming mechanism for the progesterone receptor, Nature Communications (2025). DOI: 10.1038/s41467-025-59458-y.