Uncategorized Tuesday, 2026/05/05
The findings show that the RIPK1^K376R mutant not only induces kinase activity-dependent cell death during embryonic development, but also activates the NLRP3 inflammasome in adulthood through RIPK3-mediated metabolic reprogramming, thereby promoting kinase activity-independent inflammation driven by its scaffold function.
Receptor-interacting protein kinase 1, or RIPK1, is a key regulator of cell death and inflammation, and its activity is controlled by various post-translational modifications. Although previous studies have shown that ubiquitination of lysine 376, or K376, in RIPK1 can suppress apoptosis and necroptosis both in vitro and in vivo, its role in inflammation has remained unclear.
Zhang Haibing and Li Ming from the Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, published a research paper online in PNAS as co-corresponding authors. Jianling Liu, Mingyan Xing, and Han Liu are co-first authors of the paper, titled “RIPK1 ubiquitination regulates its kinase-independent function in development and inflammation.”
In this study, the researchers introduced the kinase-inactive D138N mutation into Ripk1^K376R/K376R mice. Notably, Ripk1^K376R,D138N/K376R,D138N mice rescued the embryonic lethality observed in Ripk1^K376R/K376R mice, but developed systemic inflammation. Importantly, this inflammation was significantly alleviated when Caspase-1/11 were jointly deleted, whereas deletion of Trif had no such effect, indicating that inflammasome activation plays a key role in this process.
Mechanistically, loss of ubiquitination at the RIPK1 K376 site promoted kinase activity-dependent cell death, which forms the basis of the lethality seen in Ripk1^K376R/K376R mice. More importantly, the K376R mutation also drove inflammation independent of RIPK1 kinase activity by triggering endogenous NLRP3 inflammasome activation and downstream interleukin-1β, or IL-1β, secretion.
In addition, the authors found that RIPK1 promotes this process through a RIPK3-dependent mechanism. Consistent with this, deletion of Ripk3, but not Mlkl, improved the inflammatory phenotype, revealing an inflammatory axis independent of necroptosis.
Taken together, the findings indicate that the RIPK1^K376R mutant not only induces kinase activity-dependent cell death during embryonic development, but also activates the NLRP3 inflammasome in adulthood through RIPK3-mediated metabolic reprogramming, thereby promoting kinase activity-independent inflammation driven by the scaffold function of RIPK1.
Our Related Proteins
| Cat.No. # | Product Name | Source (Host) | Species | Tag | Protein Length | Price |
|---|---|---|---|---|---|---|
| RIPK1-202H |
Active Recombinant Human RIPK1 protein, GST-tagged
|
Insect Cells | Human | GST | 1-327 aa | |
| RIPK1-7018H |
Recombinant Full Length Human RIPK1, GST-tagged

|
Sf9 Cells | Human | GST | Full L. 1-671 a.a. | |
| RIPK1-42H | Recombinant Full Length Human RIPK1 protein, GST-tagged | Wheat Germ | Human | GST | Full L. 1-671 a.a. | |
| NLRP3-10722ME | Recombinant Mouse NLRP3 Protein, His tagged | E.coli | Mouse | His | ||
| NLRP3-3835H | Recombinant Human NLRP3 protein, His-tagged | E.coli | Human | His | 937-1036 aa | |
| NLRP3-10722M | Recombinant Mouse NLRP3 Protein | Mammalian Cells | Mouse | His |
|
|
| IL1B-1167C |
Active Recombinant Cynomolgus IL1B Protein
|
E.coli | Cynomolgus | Non | 117-269 a.a. | |
| IL1B-02H |
Recombinant Human IL1B protein
|
E.coli | Human | Non | 153 | |
| IL1B-14167H | Recombinant Human IL1B protein, GST-tagged | E.coli | Human | GST | 1-269 aa | |
| Il1b-383M | Active Recombinant Mouse Il1b protein | E.coli | Mouse | Non | Val118-Ser269 | |
| Il1b-384R | Recombinant Rat Il1b protein(Met1-Ser268), His-tagged | E.coli | Rat | His | Met1-Ser268 | |
| IL1B-577R | Recombinant Rat IL1B, His tagged | E.coli | Rat | His | 117-268 a.a. | |
| IL1B-744H | Active Recombinant Human IL1B protein, His-tagged | E.coli | Human | His | 1-269 aa | |
| IL1B-02HP | Active Recombinant Human IL1B protein, R-PE labeled | E.coli | Human | 154 aa | ||
| CASP8-26H |
Active Recombinant Human CASP8, His-tagged

|
E.coli | Human | His | 200-496 a.a. | |
| CASP8-1146R | Recombinant Rat CASP8 Protein | Mammalian Cells | Rat | His |
|
Receptor-interacting protein kinase 1, or RIPK1, is a serine/threonine kinase involved in the regulation of cell death, inflammation, and immunity. In humans, biallelic loss-of-function mutations in RIPK1 cause severe immunodeficiency and early-onset inflammatory diseases, such as inflammatory bowel disease and arthritis. In mice, deletion of the Ripk1 gene leads to perinatal death, largely due to uncontrolled apoptosis mediated by FADD/Caspase-8, abbreviated as Casp8, and necroptosis driven by the RIPK3/MLKL axis.
Although the kinase activity of RIPK1 is indispensable for RIPK1-dependent apoptosis, or RDA, and necroptosis, mice in which the kinase is genetically inactivated through kinase-dead mutations, such as D138N and K45A, show normal survival. In fact, these kinase-mutant mice have been observed to be resistant to tumor necrosis factor-α, or TNF-α-induced systemic inflammatory response syndrome, as well as multiple neurodegenerative diseases. This suggests that RIPK1 kinase activity is required not only for cell death, but also for the development of pathological inflammation.
At the same time, cleavage of RIPK1 by Casp8 serves as a critical checkpoint preventing its abnormal activation. Failure of this cleavage leads to spontaneous inflammation and death, while eliminating RIPK1 kinase activity can partially rescue this phenotype. These observations suggest that activated RIPK1 may promote inflammation through mechanisms beyond its kinase function.
Genetic studies have shown that introducing a kinase-inactivating mutation into RIPK1 completely rescues the embryonic lethality caused by the D324A mutation at the Casp8 cleavage site, but the double-mutant mice ultimately fail to survive past weaning. This indicates that activated RIPK1 mediates inflammatory functions through kinase-independent signaling pathways.
Mice with catalytically inactive Casp8 promote pyroptosis through its scaffold function, especially when apoptosis and necroptosis are inhibited. However, whether RIPK1 participates in inflammasome activation or inflammatory reprogramming through its scaffold function has remained unclear.
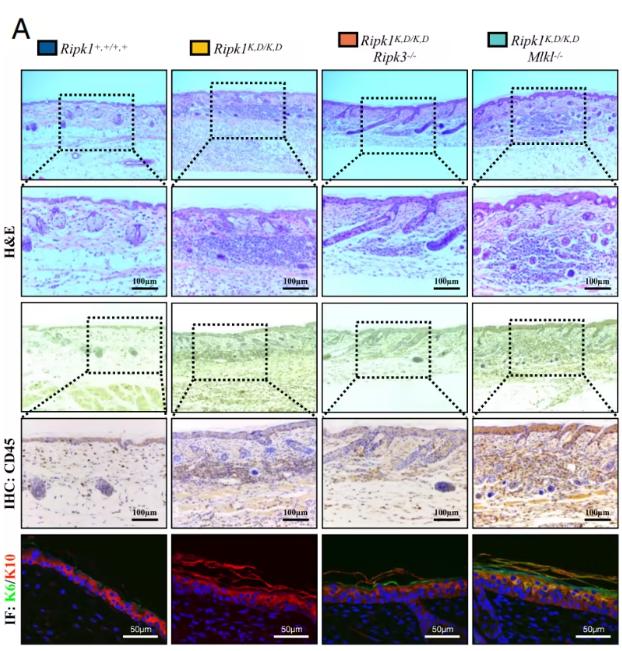
Fig1. RIPK3, rather than MLKL, is responsible for systemic inflammatory responses mediated by the RIPK1 scaffold. (Li, M., et al. PNAS)
In this study, the authors introduced the kinase-dead D138N mutation into the K376R background to generate a double-mutant mouse model, Ripk1^K376R,D138N/K376R,D138N. Strikingly, these mice rescued the embryonic lethality observed in Ripk1^K376R/K376R mice, but developed spontaneous systemic inflammation after birth. Genetic dissection showed that this inflammation could be rescued by deletion of Casp1/11, but not by deletion of TRIF, suggesting the presence of endogenous inflammasome activation.
Mechanistically, the authors found that the RIPK1 K376R mutation drives both kinase activity-dependent cell death and RIPK1 kinase activity-independent inflammation. Kinase-independent inflammation is triggered when activated RIPK1 binds to RIPK3 to reprogram arachidonic acid metabolism, leading to excessive prostaglandin production, which in turn induces endogenous NLRP3 inflammasome activation and downstream IL-1β secretion.
In summary, the study reveals that the RIPK1 K376R mutant not only promotes kinase activity-dependent cell death during embryogenesis, but also drives kinase-independent inflammatory responses after birth.
Related Products & Services
- Apoptosis Signal Pathway
- PI3K/Akt Signaling Pathway
- Immune Checkpoint Proteins
- Cancer Drug Targets
- Protein Engineering Services
- Protein Interaction Service
- Protein Expression and Purification Services
- Drug Discovery Screening
- Protein Pathway Profiling
Reference
- Li, M., Liu, J., Xing, M., Liu, H., Wang, L., Wu, X., Ou, Y., Zhao, X., Wang, Y., Xie, Y., Zhang, H., Wu, Z., Hao, J., Li, H., Li, Y., & Zhang, H. (2026). RIPK1 ubiquitination regulates its kinase-independent function in development and inflammation. Proceedings of the National Academy of Sciences, 123(15), e2520356123. https://doi.org/10.1073/pnas.2520356123