What Is Cancer Epigenetics?
Cancer epigenetics is a general term for various abnormalities related to tumors that do not involve changes in nucleic acid sequences but can cause changes in heritable gene expression. It is mainly manifested in the reduction of whole-genome DNA methylation level, hypermethylation of the promoter region, changes in specific histone modification, abnormal chromatin conformation, changes in non-coding RNA (ncRNA) molecules, etc. All of these epigenetic changes can affect the growth, immune escape, metastasis, heterogeneity and drug resistance of tumor cells, and may even contribute directly to tumor occurrence. These epigenetic abnormalities are reversible because they do not alter the properties of genomic DNA sequences, which provides a basis for the study of epigenetic drugs in tumors.
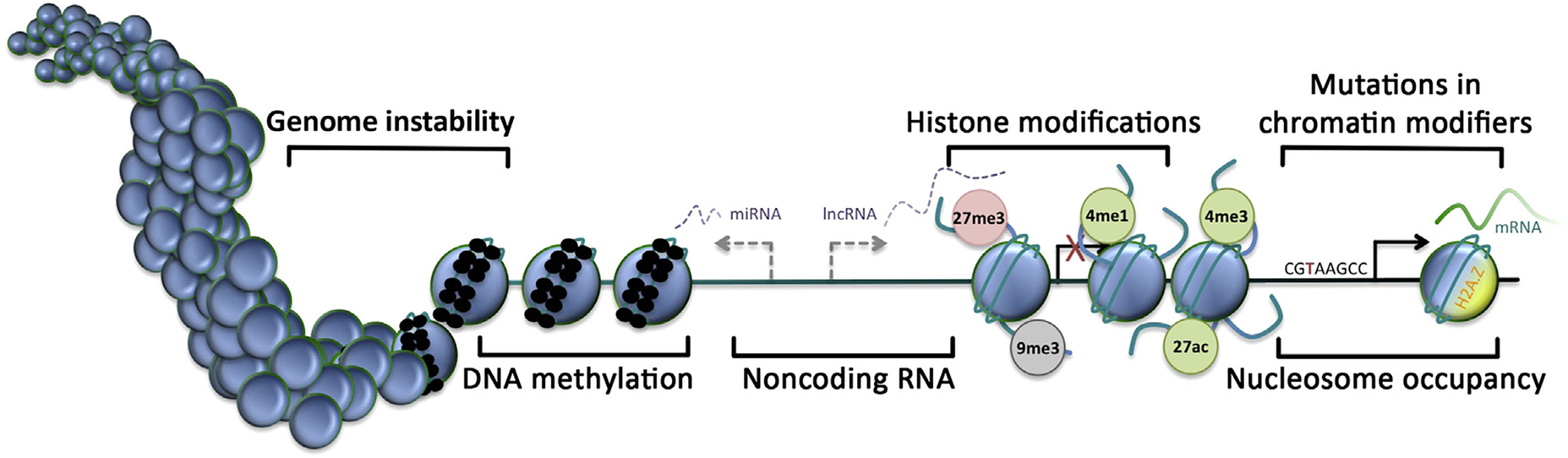
Figure 1. Epigenetic hallmarks of the cancer genome. Nucleosome, blue circle; DNA methylation, small black circle; transcriptional start site, arrow; inactive transcriptional start site, red cross; H3K27 trimethylation, 27me3 (red, repressed); H3K9 trimethylation, 9me3 (gray, silenced); H3K4 monomethylation, 4me1 (green, active); H3K4 trimethylation, 4me3 (green, active); H3K27 acetylation, 27ac (green, active).
DNA Methylation and Cancer
Changes in DNA methylation patterns are the first epigenetic event to be described in human cancer and have been observed in both early and late-stage tumors. In normal human somatic cells, DNA methylation usually occurs on dinucleotide CpG. CpG-rich regions are unevenly distributed in the genome, and they often appear in gene promoter regions. CpG sites located in the promoter region are usually not methylated, while CpG outside the regulatory region of genes is methylated. In many types of tumors, such as colon cancer, extensive hypomethylation has been observed in non-coding regions of the genome, which is thought to cause genomic instability, increase mutation rates, and may lead to aberrant activation of oncogenes. Moreover, DNA hypermethylation can also occur during the development of cancer, especially in CpG islands of the promoter region, resulting in transcriptional silencing of affected genes, such as tumor suppressor genes. Interestingly, different types of cancer exhibit different methylation patterns, so further studies are needed to clarify the differential methylation patterns of specific cancer types and the correlation of epigenomic mapping with underlying pathology.
Histone Modification and Cancer
Histone can undergo various post-translational modification (PTM) events, such as methylation, acetylation, phosphorylation, ubiquitination, etc. Histone modification will change in different biological processes, thereby providing a recognition marker and producing synergistic or antagonistic effects for the binding of other proteins to DNA. It is a dynamic transcriptional regulatory component and known as “histone code”. During the process of histone PTM, the enzymes responsible for addition or removal of these epigenetic markers are usually referred to as “writers” and “erasers”, respectively. In addition, the protein with specific reader domains that selectively recognize different histone modifications is called “readers”. The exact residue and modification type of modified histones is often associated with a panel of complementary marks that can easily be identified as “active” or “repressive” combinations.
Histone modifications related to both active and inhibitory chromatin can be observed during cancer progression, and these modifications can occur simultaneously. Changes in histone modifications can also be accompanied by other epigenetic changes, and these changes can be attributed to the aberrant activity of the “writer”, "eraser" and “reader”. Mutations in histone-modifying enzymes in tumors, such as mutations of histone methylase, histone demethylase, and histone acetylase, can also cause abnormal gene expression and determine tumor cell phenotypes.
Chromatin Remodeling and Cancer
In eukaryotic cells, chromatin remodeling factors regulate chromatin structure by altering the assembly, disassembly, and relocalization of nucleosomes, thereby improving the local accessibility of transcription-related factors in their chromatin DNA, which further initiates or suppresses transcription of related genes. Many general epigenetic changes in cancer cells are accompanied by atypical nucleosome dynamics, for example, outside of the promoter region in breast and prostate cancer cell lines, hypermethylation is accompanied by nucleosome occupancy of insulators and enhancers. In the study of Ras-induced tumor development, the importance of chromatin remodeling changes in cancer is also evident, with the observation that chromatin conformation is more open in early-developing tumors and more accessible in late-stage tumor development. It has been observed that chromatin conformation is more open in early-stage tumors and more accessible in late-stage tumors. Thus, the accessibility of chromatin may determine the aggressiveness of cancer.
Non-coding RNA (ncRNA) and Cancer
The regions of DNA that do not encode proteins account for a large portion of the human genome and these sequences are widely transcribed. In recent years, it has been found that these non-coding sequences also play a crucial role in gene regulation and cancer development. Non-coding RNAs play a role in regulating gene expression through translational repression and protein binding. More and more ncRNAs are found to be involved in cancer occurrence and development. One of the mechanisms that trigger oncogenic ncRNA changes is an epigenetic mechanism that can affect transcriptional ncRNA levels. Changes in ncRNA expression level affect target binding and therefore exert their tumorigenic effects through dysregulation of pathways of oncogene and tumor suppressor gene.
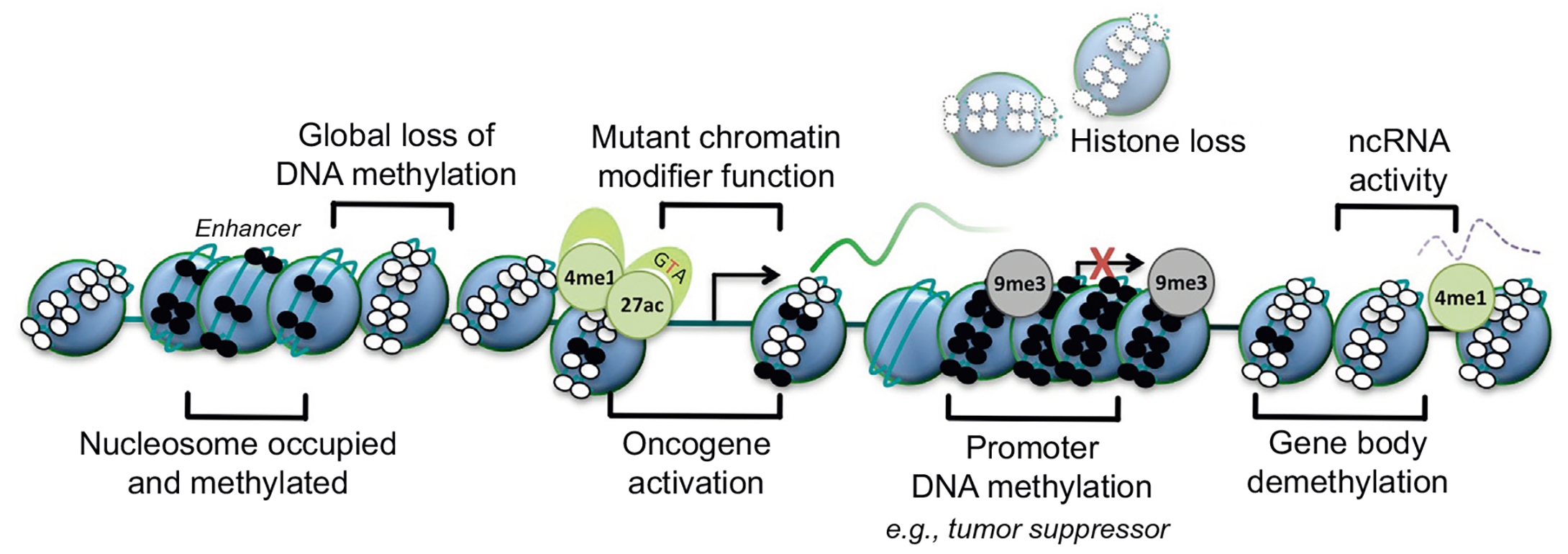
Figure 2. Basic epigenetic mechanisms in cancer. Nucleosome, blue circle; DNA methylation, small black circle; unmethylated DNA, small white circle; transcriptional start site, arrow; inactive transcriptional start site, red cross; H3K9 trimethylation, 9me3 (gray, silenced); H3K4 monomethylation, 4me1 (green, active); H3K27 acetylation, 27ac (green, active).
To promote your cancer epigenetics projects, Creative BioMart provides a variety of powerful tools (including high-quality recombinant proteins, synthetic peptides, nucleosomes, antibodies, cell lysates, stable cell lines, bioactive small molecules, and assay kits). With over 10 years’ experience, we can offer various epigenetics services, such as DNA methylation analysis, RNA methylation analysis, transcriptomics profiling, histone PTM analysis, chromatin remodeling and accessibility analysis, as well as epigenetic drug discovery.
References
1. Egger G, Arimondo P. (Eds.). Drug Discovery in Cancer Epigenetics. Academic Press, 2015.
2. Klutstein M.; et al. DNA methylation in cancer and aging. Cancer Research. 2016, 76(12): 3446-3450.
3. Audia J E, Campbell R M. Histone modifications and cancer. Cold Spring Harbor Perspectives in Biology. 2016, 8(4): a019521.
USA
Enter your email here to subscribe.
Follow us on

Easy access to products and services you need from our library via powerful searching tools