
Principle and Protocol of SDS-PAGE
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) is the most commonly used electrophoretic technique for protein separation. SDS polyacrylamide gel electrophoresis is to add SDS. SDS into the polyacrylamide gel system. The intramolecular and intermolecular chlorine bonds destroy the secondary and tertiary structure of the protein, form a negatively charged SDS protein complex, and reduce or eliminate the natural charge beauty between various protein molecules. During electrophoresis, the migration rate of protein molecules depends on the molecular size. When the relative molecular weight is 15000-20000, the mobility of the protein has a linear relationship with the logarithm of the relative molecular weight. Therefore, in addition to protein separation, this method is often used to determine the relative molecular weight of unknown proteins.
The mobility of each protein component in the protein mixed sample mainly depends on the molecular size and shape and the amount of charge. The protein loading buffer added before SDS-PAGE loading contains SDS and reducing agent DTT (or β- Mercaptoethanol), reducing agent makes protein disulfide bond open, and fold conformation is broken. SDS is an anionic surfactant. When added to the sample and electrophoresis system, it can open the ammonia bond and hydrophobic bond of the protein, and bind to the protein molecule (under certain conditions, the binding ratio of most proteins to SDS is 1.4 g SDS/1 g protein), so that all kinds of protein SDS complexes have negative charges of the same density, the amount of which is far more than the original charge of the protein molecule, Thus, the original charge difference between different kinds of proteins is masked. At this time, the electrophoretic mobility of a protein molecule is mainly determined by its relative molecular weight, while other factors can almost be ignored.
In SDS-PAGE discontinuous electrophoresis, Tris HCl buffer system is used to prepare gel buffer, with concentrated gel buffer pH6.7 and separated gel buffer pH8.9. The electrophoresis buffer solution uses Tris-glycine buffer system. In concentrated gel, the pH environment is weak acidic, so glycine is rarely dissociated, and its swimming efficiency is low under the action of electric field.
However, Cl- ions are very high, forming a zone with low conductivity between them, and protein molecules swim between them. Since the concentrated gel is a macroporous gel, it has no effect on the molecular sieve of the protein sample, and its conductivity is inversely proportional to the electric field strength. This zone forms a high voltage gradient, which enables protein molecules with almost the same charge to gather together and concentrate into a narrow zone. When the sample enters the separating gel, due to the increase of pH in the gel, it is alkaline, glycine is largely dissociated, and the swimming speed increases, which follows closely the chlorine ion. At the same time, due to the reduction of the pore size of the separating gel, it acts as a molecular sieve. Under the effect of an electric field, protein molecules are separated according to their inherent electrification and molecular size. Therefore, the influence of pH on the whole reaction system is crucial.
1. Main Instruments and Equipment
Micropipette, gel filling bracket, glass plate, comb, power supply, beaker, heater, vertical plate electrophoresis tank, DC stabilized power supply, scanner, balance.
2. Experimental Materials
Protein sample, standard protein with low relative molecular weight.
3. Main reagents
(1) Separating gel buffer (Tris-HCl buffer, pH 8.9): take 48mL of 1mol/L hydrochloric acid, 36.3g Tris, and dissolve it in non-ionized water to a constant volume of 100mL.
(2) Stacking gel buffer (Tris-HCl buffer, pH 6.7): take 48mL of 1mol/L hydrochloric acid, 5.98g Tris, and dissolve it in non-ionized water to a constant volume of 100mL.
(3) 30% Acr-Bis (Acr: Bis=29:1): Weigh 29g of acrylamide (Acr) and 1g of N, N-methylenebisacrylamide (Bis), dissolve them in double distilled water, and finally fix the volume to 100mL. After filtering, place them in a brown reagent bottle and store them at 4°C.
(4) 10% SDS solution *1: weigh 10g SDS and dissolve it in 100mL deionized water.
(5) 10% ammonium persulfate (AP): 0.1g ammonium persulfate is dissolved in 1mL H2O, and it is ready for use.
(6) Electrophoretic buffer (Tris-glycine buffer pH 8.3): weigh 6.0 g Tris, 28.8 g glycine, and 1.0 g SDS, and dissolve them in non-ionized water to a constant volume of 1 L.
(7) 2 × Sample loading buffer *2: take SDS 100 mg, β-Mercaptoethanol 0.1mL, glycerin 1mL, bromophenol blue 2mg, 0.2mol/L, pH7.2 phosphoric acid buffer 0.5mL, add double distilled water to 5mL.
(8) Staining solution: 0.25g Coomassie brilliant blue R-250, add 40mL methanol solution, 10mL glacial acetic acid and 50mL double distilled water.
(9) Decolorizing solution: 10mL glacial acetic acid, 50mL double distilled water and 40mL methanol are mixed evenly.
1. Preparation of Gel
Clean the glass plate and install the sandwich type vertical plate electrophoresis tank after it is dried. Pay attention not to touch the glass on the glue filling surface with hands during installation. Glue preparation: select the appropriate concentration of separating gel according to the relative molecular weight range of the target protein. SDS-PAGE discontinuous system is used in this experiment, and separation gel and stacking gel are prepared according to Table 2-2-2.
Table 2-2-2 Composition of SDS-PAGE
| Reagent | Separating gel (20 mL) | Stacking gel (10 mL) | |||
| 5% | 7.5% | 10% | 15% | 3% | |
| 30% Acr-Bis (40% T, 5% C) | 3.33 | 5 | 6.66 | 10 | 1 |
| Separating gel buffer (pH 8.9) | 2.5 | 2.5 | 2.5 | 2.5 | - |
| Stacking gel buffer (pH6.7) | - | - | - | - | 1.25 |
| 10% SDS | 0.2 | 0.2 | 0.2 | 0.2 | 0.1 |
| Deionized water | 11.87 | 10.2 | 8.54 | 5.2 | 5.6 |
| 10% APS*3 | 0.1 | 0.1 | 0.1 | 0.1 | 0.05 |
| TEMED*3 | 2 | 2 | 2 | 2 | 2 |
When the separating gel is poured to 3/4 of the height of the short glass plate, stop pouring gel, and add 1mL isopropanol or water for sealing. After about 30min, an obvious boundary appears between the gel and the sealing layer, indicating that the gel is fully polymerized. Pour out the sealing layer liquid, wash it with distilled water and absorb excess water with filter paper.
(1) Preparation of separating gel
20mL 10% separating gel is prepared according to the table. After mixing, use a syringe to add the gel solution into the gap between the long and short glass plates, about 8cm high. Use a 1mL syringe to take a little distilled water, slowly inject along the wall of the long glass plate, 3-4 mm high, and conduct water sealing. After about 30min, a boundary with different refractive index appears between the gel and the water seal layer, indicating that the gel is fully polymerized *4. Pour the distilled water from the water seal layer, and then absorb the excess water with filter paper.
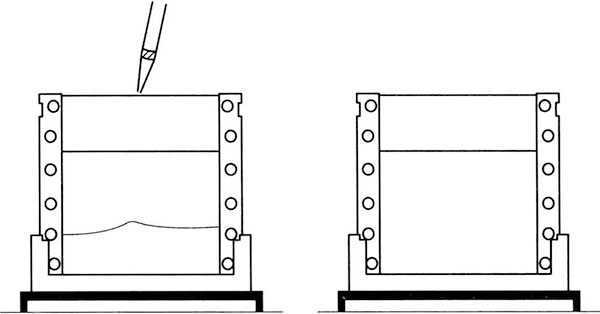
(2) Preparation of concentrated adhesive
Prepare 10mL of 3% concentrated adhesive according to the table. After mixing, use a syringe to add the concentrated adhesive to the polymerized separating adhesive until it is about 0.3cm away from the upper edge of the short glass plate. Gently insert the sample comb into the concentrated adhesive to avoid air bubbles. After about 30 minutes of gel polymerization, carefully pull out the sample comb, use narrow strip filter paper to absorb the excess water in the sample groove, pour pH8.3 Tris-glycine buffer into the upper and lower storage tanks, which should be more than 0.5cm above the short plate, and then prepare to add samples.
2. Sample Processing and Sampling*5
Each standard protein and protein to be tested are dissolved in the sample buffer solution to make the mass concentration 0.5-1 mg/mL, heated in a boiling water bath for 3 min, and cooled on ice for standby. Generally, the sample volume is 10-15 μ L (i.e. 2-10 μg protein). If the sample is thin, the sample volume can be increased. Use a micropipette to carefully add the sample to the bottom of the concave sample hole of the gel through the buffer. After all the concave sample holes are filled with samples, electrophoresis can begin.
3. Electrophoresis
Turn on the switch of the DC stabilized voltage electrophoresis apparatus to start electrophoresis*6. When the blue dye moves to 1cm from the bottom, turn off the power. Pull out the fixed plate, take out the glass plate, gently pry off a piece of glass with a plastic pry board, cut a corner at one end of the rubber plate as a mark, and move the rubber plate to a plastic box for dyeing.
4. Dyeing and Decolorization
Pour the dye solution into the plastic box, dye it for about 30 min, rinse it with distilled water for several times, and then decolorize it with the decolorizing solution until the protein zone is clear. Scan and record the data with a scanner.
1. Concentration of SDS monomer in solution: SDS exists in aqueous solution as a mixture of monomer and SDS polypeptide micelle, and it is monomer that can combine with protein molecules. In order to ensure the full combination of protein and SDS, their mass ratio should be 1:4 or 1:3.
2. The amount of SDS bound to protein only depends on the concentration of SDS monomer at equilibrium, not the total concentration. Only in the solution with low ionic strength, SDS monomer has a high equilibrium concentration. Therefore, the sample buffer ionic strength of SDS electrophoresis is low, usually 10-100mmol/L.
3. Only after the disulfide bond is completely reduced can the protein molecules be depolymerized, and SDS can be quantitatively bound to the subunits to give the linear relationship between the relative mobility and the logarithm of the relative molecular mass. The content of β-mercaptoethanol in the sample buffer is usually 4% - 5%, and the content of dithiothreitol is usually 2% - 3%.
4. When SDS-PAGE gel electrophoresis is used to determine the relative molecular weight of proteins, the relative molecular weight of proteins with abnormal charge or conformation, proteins with large auxiliary groups (such as some glycoproteins) and some structural proteins such as collagen are unreliable. For example, histone H1 itself has a large amount of positive charges. Therefore, although SDS in normal proportion is combined, the influence of its original positive charges cannot be completely covered. Its relative molecular weight is 2.1 × 104, but the result of SDS-PAGE gel electrophoresis is 3.5 × 104。 Therefore, it is better to use at least two methods to determine the relative molecular weight of unknown samples for mutual verification.
5. It should be noted that poor sample melting effect or excessive concentration of separating gel will lead to the tailing of electrophoresis bands.
*1 It is easy to precipitate and crystallize at low temperature. It is heated slightly before use to make it completely dissolved. Functions: remove protein charges, dissociate hydrogen bonds between proteins, cancel hydrophobic interactions within protein molecules, and remove polypeptide folding.
*2 Restore SDS processing. Add SDS and DTT (or β- Mercaptoethanol), the protein conformation is dissociated, the charge is neutralized, and the SDS protein binding molecule is formed. In electrophoresis, the separation is only based on the relative molecular weight. Generally, electrophoresis is handled in this way. The sample is diluted to an appropriate concentration, centrifuged by adding sample buffer, boiled in boiling water for 5min, and centrifuged for sample addition.
*3 Add before pouring glue.
*4 The glue needs to be fully solidified. If the middle part of the gel is not uniformly solidified, a "smile" shaped belt will be formed (both sides are raised and the middle is concave).
*5 Reduced SDS treatment, non-reduced SDS treatment and reduced SDS treatment with alkylation are selected according to the different purposes of sample separation. Non reducing SDS treatment: physiological body fluid, serum, urine and other samples are generally boiled in 1% SDS boiling water for 3min without adding reducing agent. Reduced SDS treatment with alkylation: alkylation of amine iodoacetate can protect - SH group well and firmly, and obtain narrow spectral band. In addition, iodoacetate can trap excessive DTT and prevent the texture phenomenon in silver staining. one hundred μL sample buffer contains 10 μL 20% iodoacetate amine and keep it at room temperature for 30min.
*6 Adjust the voltage to 80V when using MiniVE glue tank. When the sample enters the separating gel, adjust the voltage to 120V.