


Apoptosis Adaptor Proteins
Related Symbol Search List
- BCAP31
- CALD1
- CD2AP
- CIDEA
- CIDEC
- CRADD
- CRKL
- DFFA
- FADD
- MFGE8
- MYD88
- PEA15
- PYCARD
- RIPK1
- RALBP1
- MPRIP
- RIPK3
- sfn
- TANK
- TICAM2
- TRADD
- TRAF1
- 14-3-3 beta
- 14-3-3 epsilon
- YWHAG
- YWHAH
- YWHAQ
- YWHAZ
Immunology Background
Background
Apoptosis adaptor proteins play a pivotal role in both the intrinsic and extrinsic pathways of apoptosis, acting as critical molecular intermediaries that ensure the precise and timely execution of the cell death program. These proteins act as molecular bridges that link upstream apoptotic signals to downstream activation of caspases, the proteolytic enzymes responsible for cell degradation.
Structure and Function of Apoptosis Adaptor Proteins
Apoptosis adaptor proteins are typically characterized by the presence of specific protein-protein interaction domains that enable them to bind to other proteins and facilitate the formation of signaling complexes. The most well-known domains involved in apoptosis adaptor function include the death domain (DD), the death effector domain (DED), the caspase recruitment domain (CARD), and the Bcl-2 homology domain (BH3).
Death Domain (DD)
The death domain is a protein interaction module found in many apoptosis-related proteins, including adaptor proteins such as Fas-associated death domain (FADD) and tumor necrosis factor receptor type 1-associated death domain (TRADD). The DD allows these adaptors to interact with death receptors and other proteins containing similar domains, forming a signaling complex that leads to the recruitment and activation of initiator caspases, such as caspase-8 in the extrinsic pathway.
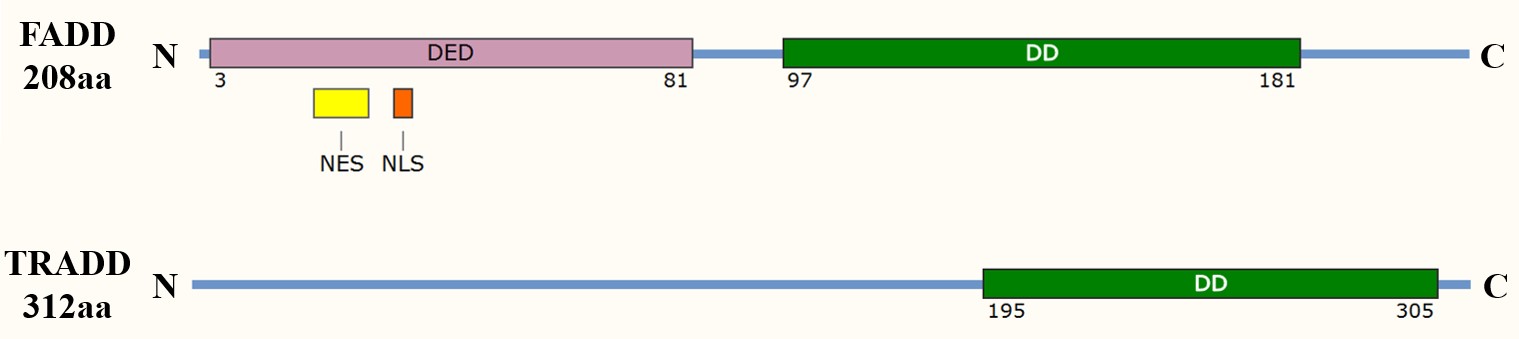 Fig. 1: Domain organization of the apoptosis adaptor proteins FADD and TRADD. DED: death effector domain; DD: death domain; NES: nuclear export signal; NLS: nuclear localization sequence.
Fig. 1: Domain organization of the apoptosis adaptor proteins FADD and TRADD. DED: death effector domain; DD: death domain; NES: nuclear export signal; NLS: nuclear localization sequence.Death Effector Domain (DED)
The DED is another protein interaction domain that mediates interactions between adaptor proteins and caspases. For example, FADD contains a DED that allows it to bind to procaspase-8, facilitating its activation. The DED is also found in proteins such as cFLIP, which can inhibit apoptosis by competing with caspase-8 for binding to FADD, thereby regulating the sensitivity of cells to apoptotic signals.
Caspase Recruitment Domain (CARD)
The CARD is a domain that mediates interactions between caspases and adaptor proteins in the intrinsic pathway of apoptosis. Apoptosis protease-activating factor 1 (Apaf-1), an essential adaptor protein in the intrinsic pathway, contains a CARD that allows it to bind to and recruit procaspase-9 to the apoptosome, a multi-protein complex that forms in response to cytochrome c release from the mitochondria. This recruitment is critical for the activation of caspase-9, which then activates downstream effector caspases such as caspase-3.
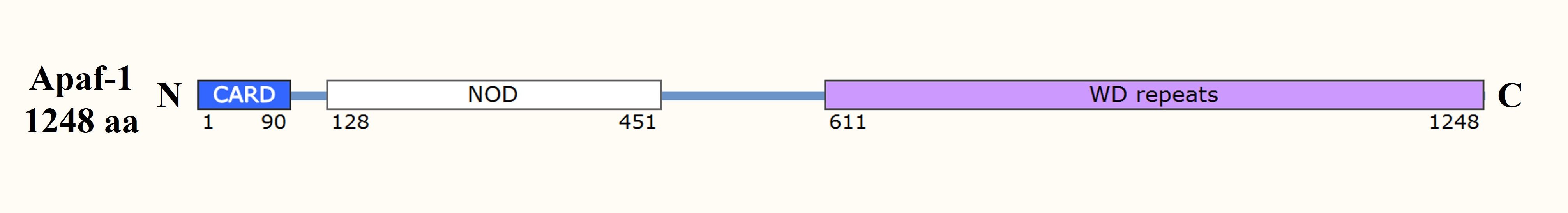 Fig. 2: Domain organization of the apoptosome component Apaf-1. CARD: caspase recruitment domain; NOD: nucleotide-binding and oligomerization domain.
Fig. 2: Domain organization of the apoptosome component Apaf-1. CARD: caspase recruitment domain; NOD: nucleotide-binding and oligomerization domain.Bcl-2 Homology Domain (BH3)
The BH3 domain is found in pro-apoptotic members of the Bcl-2 family, such as Bim, Bid, and Bad, which act as apoptosis adaptors by interacting with and neutralizing anti-apoptotic Bcl-2 proteins. This interaction promotes the release of cytochrome c from the mitochondria, triggering the intrinsic apoptotic pathway.
Specifically, Bcl-2 is a member of a large family of proteins involved in both the prevention and promotion of apoptosis. In addition to their common BH3 domain, some, like Bcl-2 itself, have the additional BH1 and BH2 domains. Several also have transmembrane (TM) domains for membrane anchoring and BH4 domains that function to inhibit apoptosis, in part by blocking the release of calcium ions from the endoplasmic reticulum. The pro-apoptotic proteins are grouped into the Bax family and have several domains homologous to domains of Bcl-2, whereas the "BH3-only" proteins have only the BH3 domain in common with Bcl-2. Members of the Bax family normally reside in an inactive form in the outer mitochondrial membrane or in the cytosol, whereas inactive members of the BH3-only family are normally restricted to the cytosol; both classes of proteins are activated and/or translocated to the mitochondria by pro-apoptotic signals.
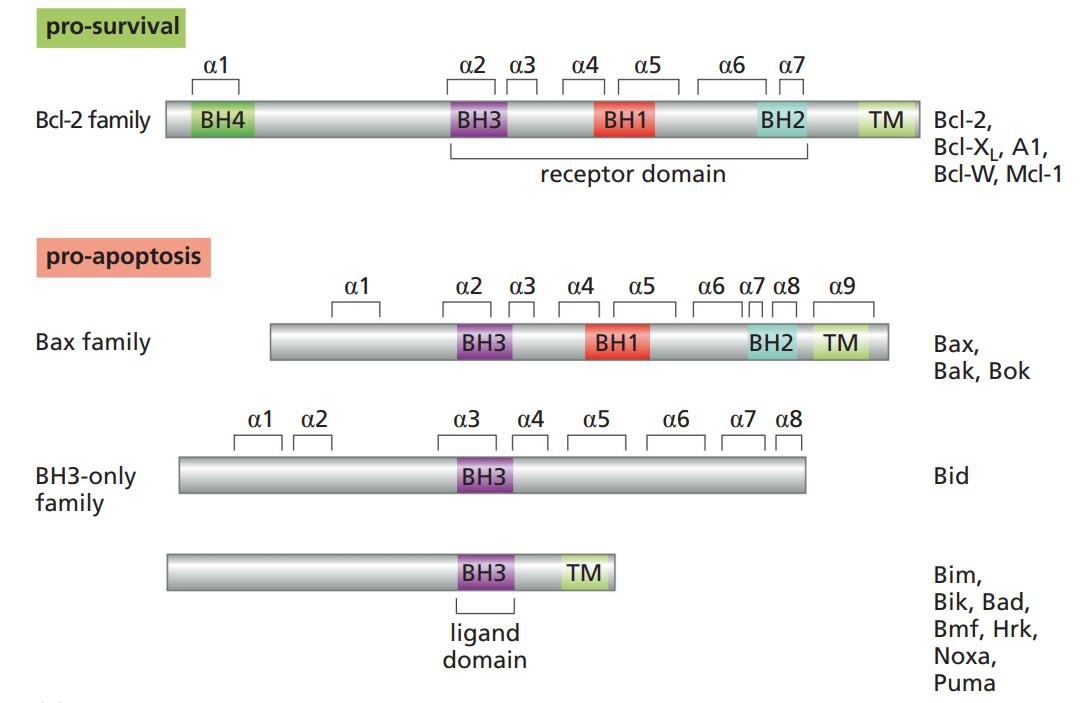 Fig. 3: Bcl-2 and related proteins (The Biology of Cancer, Garland Science, 2nd Edition).
Fig. 3: Bcl-2 and related proteins (The Biology of Cancer, Garland Science, 2nd Edition).Role of Apoptosis Adaptor Proteins in the Extrinsic Pathway
In the extrinsic pathway, apoptosis adaptor proteins are key players in the formation of the death-inducing signaling complex (DISC), a multi-protein complex that forms upon activation of death receptors by their respective ligands. The best studied example of this pathway is the Fas receptor (also known as CD95 or Apo-1) and its ligand, FasL.
Upon binding of FasL to Fas, the receptor undergoes oligomerization, leading to the recruitment of the adaptor protein FADD via interactions between their respective death domains. FADD, in turn, recruits procaspase-8 via its death effector domain, leading to the formation of the DISC. Within this complex, procaspase-8 undergoes dimerization and activation, leading to its cleavage into the active enzyme. Active caspase-8 can then cleave and activate downstream effector caspases, such as caspase-3, or cleave the BH3-only protein Bid, linking the extrinsic pathway to the intrinsic pathway.
Regulation of DISC assembly and function is critical for controlling the sensitivity of cells to apoptotic signals. Several proteins, including the cellular FLICE inhibitory protein (cFLIP), can inhibit DISC formation by competing with procaspase-8 for binding to FADD, thereby blocking activation of the extrinsic pathway. The balance between pro- and anti-apoptotic signals at the DISC level determines whether a cell will undergo apoptosis in response to extrinsic cues.
Role of Apoptosis Adaptor Proteins in the Intrinsic Pathway
In the intrinsic pathway, apoptosis adaptor proteins are involved in the formation of the apoptosome, a multi-protein complex that forms in response to mitochondrial outer membrane permeabilization (MOMP) and the release of cytochrome c into the cytosol. The apoptosome serves as a platform for the activation of procaspase-9, which then activates downstream effector caspases, such as caspase-3, leading to cell death.
The formation of the apoptosome is initiated by the release of cytochrome c from the mitochondria, a process regulated by members of the Bcl-2 family of proteins. Once in the cytosol, cytochrome c binds to Apaf-1, an adaptor protein that undergoes conformational changes and oligomerizes into a heptameric structure known as the apoptosome. Apaf-1 contains a CARD that interacts with the CARD of procaspase-9, leading to its recruitment to the apoptosome and subsequent activation.
The activity of the apoptosome is tightly regulated by several factors, including the availability of cytochrome c, the presence of inhibitor of apoptosis proteins (IAPs), and the expression of pro- and anti-apoptotic Bcl-2 family members. Apoptosis adaptor proteins such as Apaf-1 thus play a central role in determining the cell's response to intrinsic apoptotic signals by facilitating the formation and function of the apoptosome.
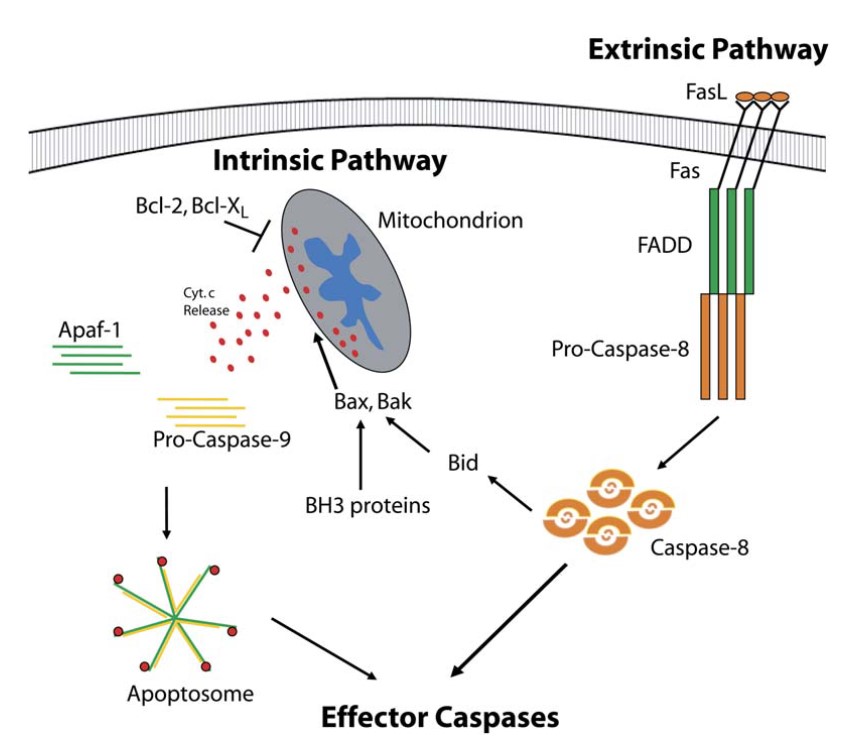 Fig. 4: Apoptosis adaptor proteins are involved in both intrinsic and extrinsic apoptotic pathways (Schafer and Kornbluth, 2006).
Fig. 4: Apoptosis adaptor proteins are involved in both intrinsic and extrinsic apoptotic pathways (Schafer and Kornbluth, 2006).Clinical Significance and Therapeutic Potential
Dysregulation of apoptosis has been implicated in a wide range of diseases, including cancer, autoimmune disorders, and neurodegenerative diseases. In cancer, for example, overexpression of anti-apoptotic proteins or loss of pro-apoptotic signals can lead to uncontrolled cell proliferation and resistance to therapy. Conversely, excessive apoptosis contributes to the pathogenesis of neurodegenerative diseases such as Alzheimer's and Parkinson's, where neuronal loss is associated with abnormal activation of apoptotic pathways.
The central role of apoptosis adaptor proteins in the regulation of cell death makes them attractive targets for therapeutic intervention. In cancer, strategies aimed at reactivating apoptosis in tumor cells by targeting adaptor proteins or their interactions are being explored. For example, small molecule inhibitors that mimic the action of pro-apoptotic BH3-only proteins, known as BH3 mimetics, such as ABT-737, Navitoclax (ABT-263) (Fig. 5) and gossypol, have shown promise in inducing apoptosis in cancer cells by neutralizing anti-apoptotic Bcl-2 proteins and promoting activation of the intrinsic pathway.
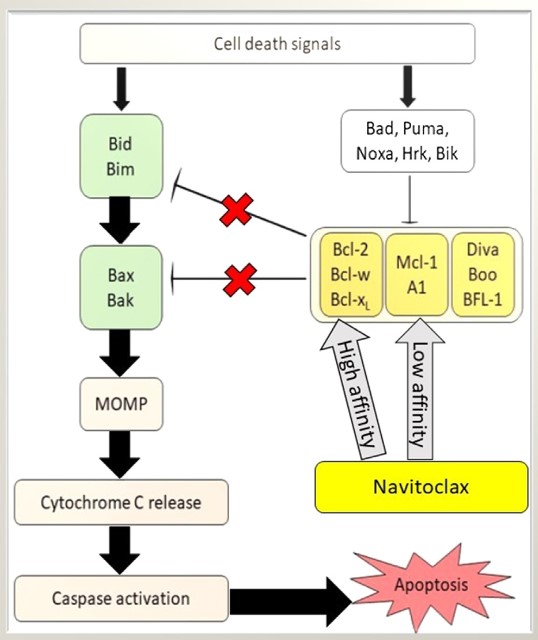 Fig. 5: Potentiation of apoptotic activity in the presence of Navitoclax. The affinity of Navitoclax for antiapoptotic Bcl-2 proteins is variable. MOMP, mitochondrial outer membrane permeabilization (Mohamad Anuar et al., 2020).
Fig. 5: Potentiation of apoptotic activity in the presence of Navitoclax. The affinity of Navitoclax for antiapoptotic Bcl-2 proteins is variable. MOMP, mitochondrial outer membrane permeabilization (Mohamad Anuar et al., 2020).In addition, the development of therapeutic agents that can modulate the activity of adaptor proteins involved in the extrinsic pathway, such as FADD or TRADD, has the potential to enhance the efficacy of immune-based therapies that rely on death receptor activation to induce tumor cell death. Similarly, strategies aimed at inhibiting the aberrant activation of apoptotic pathways in neurodegenerative diseases may offer new avenues for neuroprotection and preservation of neuronal function.
Case Study
Case 1: Tse, C.; et al. A potent and orally bioavailable bcl-2 family inhibitor. Cancer Research, 2008; 68(9), 3421–3428.
ABT-737 is a potent, small molecule inhibitor of the Bcl-2 family of proteins that is not orally bioavailable, which would limit chronic single agent therapy and dosing flexibility in combination regimens. This study evaluated the oral bioavailability of ABT-263, a Bad-like BH3 mimetic. ABT-263 disrupts Bcl-2/Bcl-xL interactions with pro-death proteins (e.g. Bim), leading to the induction of apoptosis within 2 hours of treatment. Oral administration of ABT-263 alone induces complete tumor regression in xenograft models of small cell lung cancer and acute lymphoblastic leukemia.
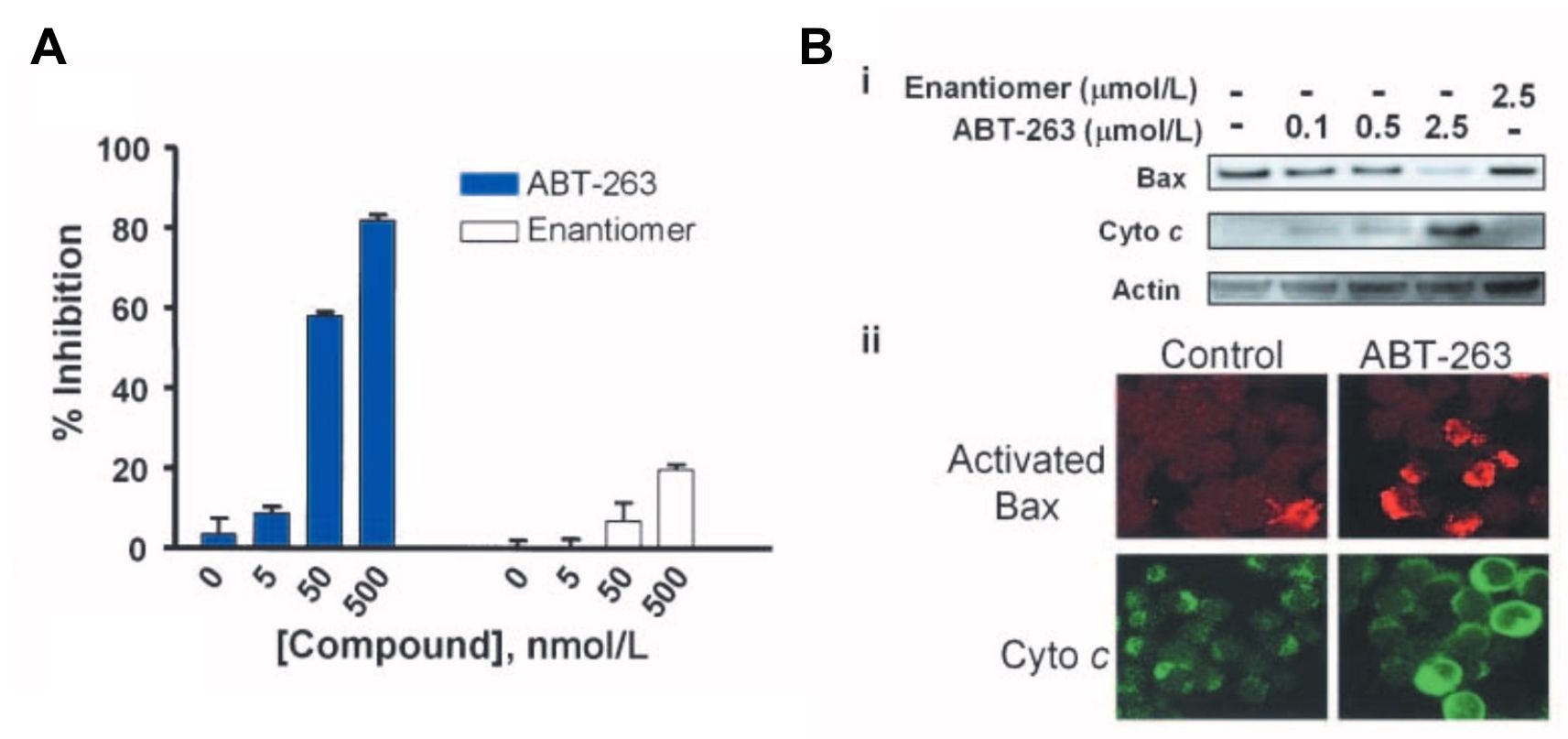 Fig. 6: Inhibition of the antiapoptotic Bcl-2 family proteins by ABT-263 induces mitochondria-dependent apoptosis. A, disruption of Bcl-xL/Bcl-xS interactions assessed by the mammalian two-hybrid system in HeLa cells. Cells were treated with 0 to 500 nmol/L of ABT-263 (closed columns) or the enantiomer (open columns) and disruption was assessed with BrightGlo. Columns, mean (n = 3); bars, SD. B, activation of Bax and cytochrome c (Cyto c) release on inhibition of Bcl-2 and Bcl-xL in H146 cells. i, immunoblots of Bax and cytochrome c in cytosolic fractions isolated 2 h posttreatment with various concentrations of ABT-263 or the enantiomer. ii, immunohistochemistry of cells with anti–activated Bax (red) and anti– cytochrome c (green) antibodies.
Fig. 6: Inhibition of the antiapoptotic Bcl-2 family proteins by ABT-263 induces mitochondria-dependent apoptosis. A, disruption of Bcl-xL/Bcl-xS interactions assessed by the mammalian two-hybrid system in HeLa cells. Cells were treated with 0 to 500 nmol/L of ABT-263 (closed columns) or the enantiomer (open columns) and disruption was assessed with BrightGlo. Columns, mean (n = 3); bars, SD. B, activation of Bax and cytochrome c (Cyto c) release on inhibition of Bcl-2 and Bcl-xL in H146 cells. i, immunoblots of Bax and cytochrome c in cytosolic fractions isolated 2 h posttreatment with various concentrations of ABT-263 or the enantiomer. ii, immunohistochemistry of cells with anti–activated Bax (red) and anti– cytochrome c (green) antibodies.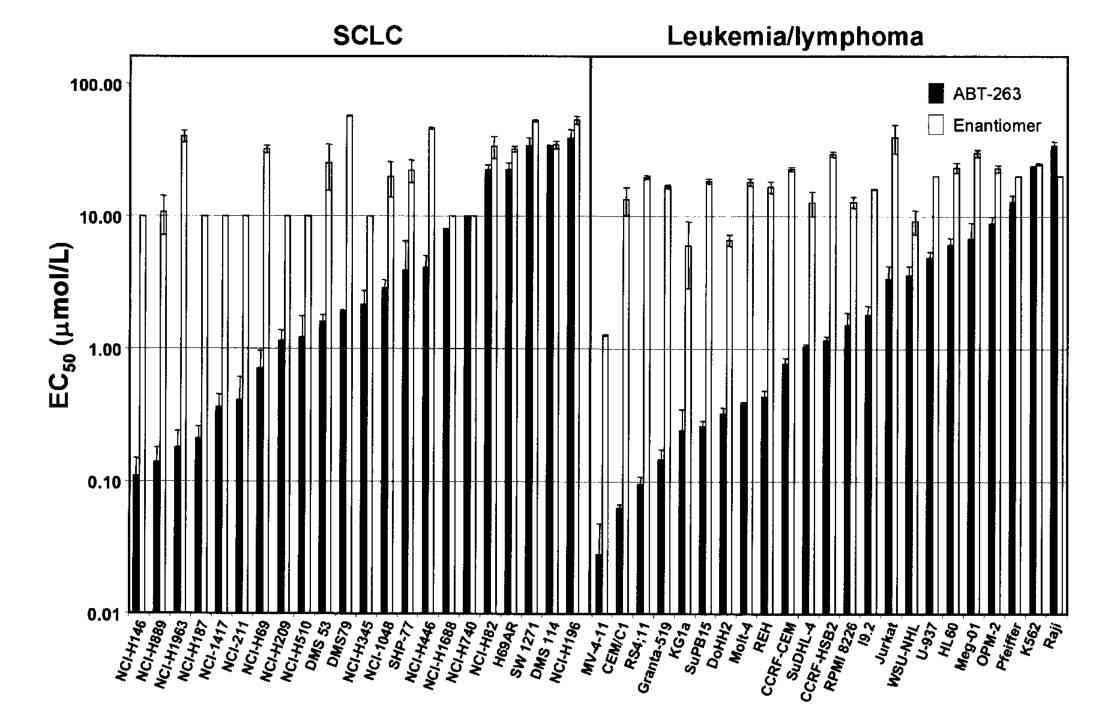 Fig. 7: Cellular activity of ABT-263 in vitro. EC50 values of ABT-263 (closed columns) or the enantiomer (open columns) against a panel of SCLC (left) or leukemia/ lymphoma (right) human tumor cell lines in the presence of 10% human serum. Columns, mean (n≥3); bars, SD.
Fig. 7: Cellular activity of ABT-263 in vitro. EC50 values of ABT-263 (closed columns) or the enantiomer (open columns) against a panel of SCLC (left) or leukemia/ lymphoma (right) human tumor cell lines in the presence of 10% human serum. Columns, mean (n≥3); bars, SD.Case 2: Schinske, K. A.; et al. A novel kinase inhibitor of fadd phosphorylation chemosensitizes through the inhibition of nf-κb. Molecular Cancer Therapeutics, 2011; 10(10), 1807–1817.
Evidence suggests that elevated levels of phosphorylated FADD in tumor cells are associated with increased activation of the anti-apoptotic transcription factor NF-κB, serving as a biomarker for aggressive disease and poor clinical outcomes. These findings suggest that targeting FADD phosphorylation may be a promising strategy for cancer therapy. A high-throughput screen using a cell-based assay to monitor FADD kinase activity identified NSC 47147 as a small molecule inhibitor of FADD phosphorylation. This compound was evaluated in live cells and mouse tumors for its ability to inhibit FADD kinase activity by targeting CK1α. NSC 47147 was shown to reduce phosphorylated FADD levels and NF-κB activity, making it a potential candidate for use in combination cancer therapy.
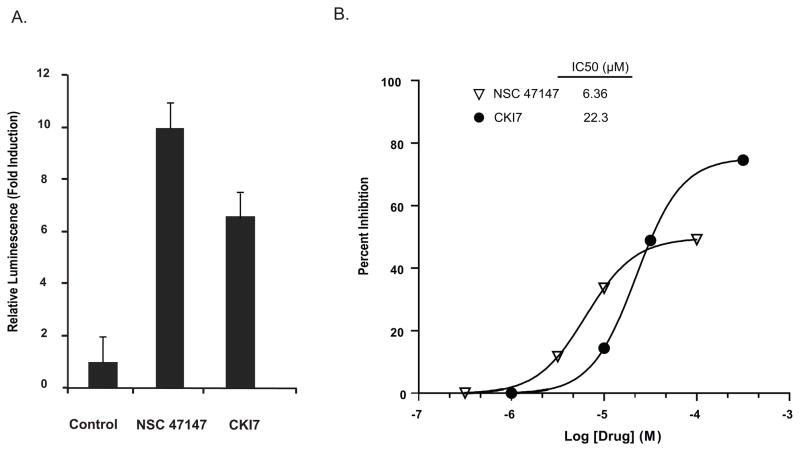 Fig. 8: NSC 47147 inhibits CK1α. (A) SW620 CK1α reporter cells were treated with 3 μM NSC 47147 and 250 μM CKI7 for 3 hours. Data shows 10-fold increase in bioluminescence indicative of CK1α inhibition in response to NSC 47147 treatment. CKI7, a CK1α inhibitor, is shown as a positive control. (B) NSC 47147 was evaluated using a TR-FRET biochemical assay for its direct effect on CK1α enzymatic activity. The in vitro assay reveals direct inhibition of CK1α activity by NSC 47147.
Fig. 8: NSC 47147 inhibits CK1α. (A) SW620 CK1α reporter cells were treated with 3 μM NSC 47147 and 250 μM CKI7 for 3 hours. Data shows 10-fold increase in bioluminescence indicative of CK1α inhibition in response to NSC 47147 treatment. CKI7, a CK1α inhibitor, is shown as a positive control. (B) NSC 47147 was evaluated using a TR-FRET biochemical assay for its direct effect on CK1α enzymatic activity. The in vitro assay reveals direct inhibition of CK1α activity by NSC 47147.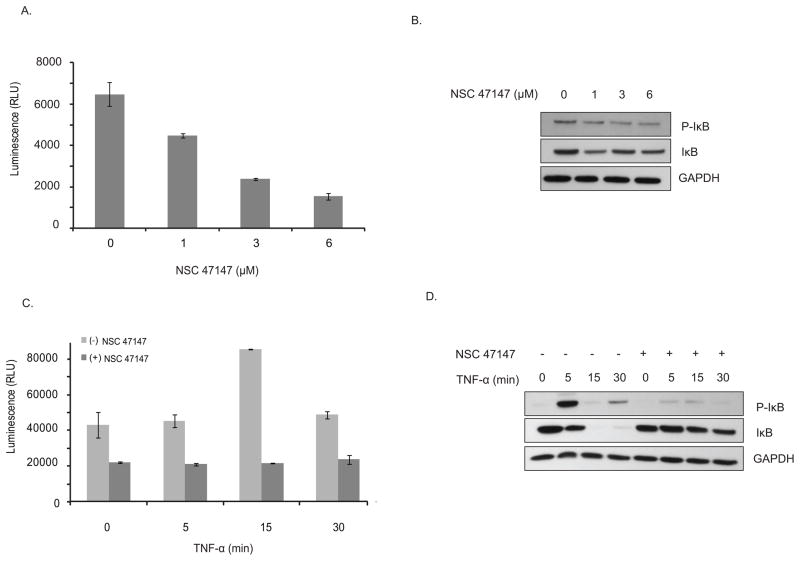 Fig. 9: NSC 47147 attenuation of NF-κB activity (A) The phosphorylation status of I-κBα was evaluated using AlphaScreen pIκB assay and by (B) western blot analysis in A549 cells following 6-hour treatment with 0, 1, 3, and 6 μM NSC 47147. Results show a decrease in luminescence indicative of pIκB inhibition. Western blot confirms decreasing levels of pIκB proteins and total I-κBα proteins in response to NSC 47147. (C) TNFα induced pIκB levels after 6-hour treatment with and without NSC 47147. A549 cells were stimulated with 10 ng/ml TNFα following pre-treatment with 6 μM NSC 47147. Cellular lysates were subjected to analysis by pIκB AlphaScreen and (D) western blot.
Fig. 9: NSC 47147 attenuation of NF-κB activity (A) The phosphorylation status of I-κBα was evaluated using AlphaScreen pIκB assay and by (B) western blot analysis in A549 cells following 6-hour treatment with 0, 1, 3, and 6 μM NSC 47147. Results show a decrease in luminescence indicative of pIκB inhibition. Western blot confirms decreasing levels of pIκB proteins and total I-κBα proteins in response to NSC 47147. (C) TNFα induced pIκB levels after 6-hour treatment with and without NSC 47147. A549 cells were stimulated with 10 ng/ml TNFα following pre-treatment with 6 μM NSC 47147. Cellular lysates were subjected to analysis by pIκB AlphaScreen and (D) western blot.References
- Mohamad Anuar, N. N., Nor Hisam, N. S., Liew, S. L., & Ugusman, A. (2020). Clinical review: Navitoclax as a pro-apoptotic and anti-fibrotic agent. Frontiers in Pharmacology, 11, 564108.
- Park, H. H. (2012). Structural features of caspase-activating complexes. International Journal of Molecular Sciences, 13(4), 4807–4818.
- Schafer, Z. T., & Kornbluth, S. (2006). The apoptosome: Physiological, developmental, and pathological modes of regulation. Developmental Cell, 10(5), 549–561.
- Schinske, K. A., Nyati, S., Khan, A. P., Williams, T. M., Johnson, T. D., Ross, B. D., Pérez Tomás, R., & Rehemtulla, A. (2011). A novel kinase inhibitor of fadd phosphorylation chemosensitizes through the inhibition of nf-κb. Molecular Cancer Therapeutics, 10(10), 1807–1817.
- Townsend, P. A., Kozhevnikova, M. V., Cexus, O. N. F., Zamyatnin, A. A., & Soond, S. M. (2021). BH3-mimetics: Recent developments in cancer therapy. Journal of Experimental & Clinical Cancer Research, 40(1), 355.
- Tse, C., Shoemaker, A. R., Adickes, J., Anderson, M. G., Chen, J., Jin, S., Johnson, E. F., Marsh, K. C., Mitten, M. J., Nimmer, P., Roberts, L., Tahir, S. K., Xiao, Y., Yang, X., Zhang, H., Fesik, S., Rosenberg, S. H., & Elmore, S. W. (2008). Abt-263: A potent and orally bioavailable bcl-2 family inhibitor. Cancer Research, 68(9), 3421–3428.