


Targets of CAR-T Cell Therapy
🧪 GPC3-2551H
Source: HEK293
Species: Human
Tag: His
Conjugation:
Protein Length: 25-559 aa


🧪 EGFR-30H
Source: Human Cells
Species: Human
Tag: His
Conjugation:
Protein Length: 25-645 aa
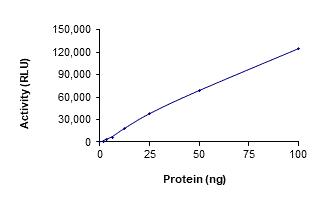
🧪 EPCAM-81H
Source: HEK293
Species: Human
Tag: Fc
Conjugation:
Protein Length: 24-265 aa

🧪 CD33-459H
Source: HEK293
Species: Human
Tag: Fc
Conjugation:
Protein Length:
🧪 CD274-592H
Source: Human Cells
Species: Human
Tag: His
Conjugation:
Protein Length: 1-239 aa

🧪 Cd274-593M
Source: Human Cells
Species: Mouse
Tag: Fc&His
Conjugation:
Protein Length: 1-238 aa
🧪 CA9-256H
Source: HEK293
Species: Human
Tag: His
Conjugation:
Protein Length: Gln138-Asp414
🧪 TNFRSF8-642H
Source: HEK293
Species: Human
Tag: Fc&His
Conjugation:
Protein Length: 1-379 aa

🧪 IL1RAP-175H
Source: Human Cells
Species: Human
Tag: Fc&His
Conjugation:
Protein Length: 1-359 aa

🧪 EGFR-692H
Source: HEK293
Species: Human
Tag: Fc
Conjugation:
Protein Length: 1-640 aa
🧪 Il13ra2-764M
Source: HEK293
Species: Mouse
Tag: Fc&His
Conjugation:
Protein Length: Met1-Lys334
🧪 IL13RA2-765H
Source: HEK293
Species: Human
Tag: Fc&His
Conjugation:
Protein Length: Met1-Leu342
🧪 PDCD1-822H
Source: HEK293
Species: Human
Tag: His
Conjugation:
Protein Length: Met 1-Gln 167
🧪 Pdcd1-823M
Source: HEK293
Species: Mouse
Tag: His
Conjugation:
Protein Length: Met1-Gln167
🧪 ERBB2-922H
Source: HEK293
Species: Human
Tag: His
Conjugation:
Protein Length: 23-652 aa

Background
What is CAR-T therapy?
For years, the foundations of cancer treatment were surgery, chemotherapy, and radiation therapy. But over the past several years, immunotherapy-therapies that enlist and strengthen the power of a patient's immune system to attack tumors-has emerged as what many in the cancer community now call the "fifth pillar" of cancer treatment.
T cells are immune system cells that play several key roles in the body's fight against the disease. They help the immune system respond to disease and directly kill diseased cells.
Unfortunately, naturally occurring T cells are not good at recognizing and fighting cancer cells.
A rapidly emerging immunotherapy approach is called adoptive cell transfer (ACT). ACT is defined as collecting and using the patients' own immune cells to treat their cancer. There are several types of ACT (see "ACT: TILs, TCRs, and CARs"), but thus far, the one that has advanced the furthest in clinical development is called CAR T-cell therapy.
Chimeric antigen receptors (CARs, also known as chimeric immunoreceptors, chimeric T cell receptors, artificial T cell receptors or CAR-T) are engineered receptors that graft an arbitrary specificity onto T cell, these receptors are used to graft the specificity of a monoclonal antibody onto a T cell, with the transfer of their coding sequence facilitated by retroviral vectors. The receptors are called chimeric because they are composed of parts from different sources.
CAR T-cell therapy is a type of immunotherapy that changes a patients’ own T cells so that they are able to recognize and attack cancer. T cells are taken from a patient's blood. Then the gene for a special receptor that binds to a certain protein on the patient's cancer cells is added in the laboratory.
Which cancers can be treated with CAR-T cell therapy?
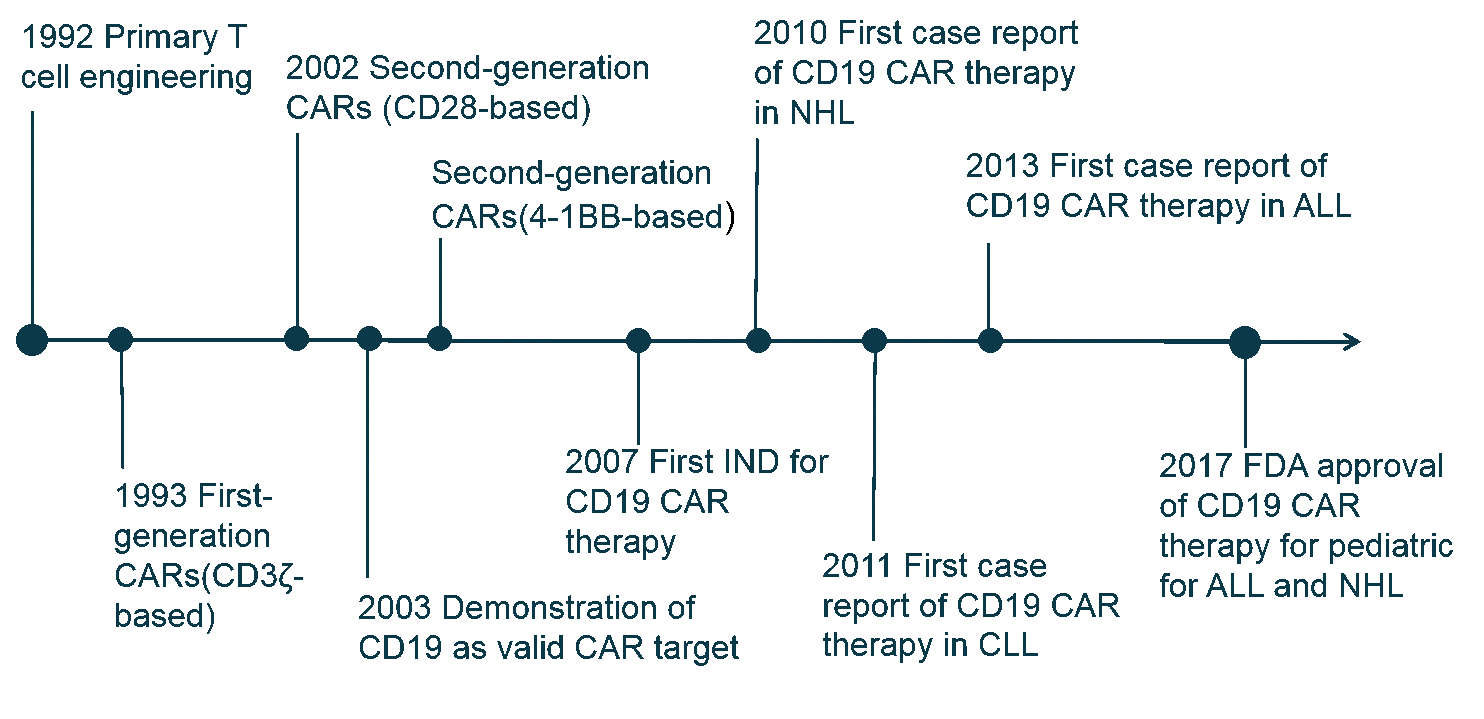
After years of clinical trials, in 2017, the Food and Drug Administration approved the use of two CAR T-cell therapies. Both use genetically modified cells that recognize a protein (called CD-19) on the surface of cancerous B cells.
- Yescarta has been approved for patients with large B-cell lymphoma that has relapsed or does not respond to standard treatments.
- Kymriah is for pediatric and young adult patients age 25 or younger with B-cell acute lymphoblastic leukemia.

To be eligible for either treatment, patients must have been treated unsuccessfully with at least two other cancer therapies.
CAR-T cells are more effective on hematologic tumors. There is some skepticism that CAR-T cells will have the same success in solid tumors. Efforts to identify unique antigens on the surface of solid tumors have largely been unsuccessful. Researchers estimate that the overwhelming majority of tumor antigens reside inside tumor cells, meaning that they are out of reach for CAR-T cells because CAR-T cells can only bind to antigens on the cell surface. Many tumor-associated antigens were already targeted to get the optimal efficacy, such as epidermal growth factor receptor (EGFR) and EGFR variant (EGFRvIII), Interleukin 13 receptor α2 (IL13Rα2), HER2, and so on. Early reports from these trials have not reported the same success that has been seen with blood cancers.
Another key obstacle to solid tumors is that components of the microenvironment that surrounds them conspire to blunt the immune response. T cells must be able to traffic to tumor sites in order to exert their effector functions in vivo. The extracellular matrix (ECM) is the main barrier to transport because it contains the heparan sulfate proteoglycans (HSPGs), the main component of ECM. T cells must get rid of HSPGs in the stroma-rich tumor microenvironment to reach tumor sites. However, T cells do not have the ability to express the enzyme heparanase (HPSE) to degrade heparan sulfate proteoglycans. Therefore, CAR-T cells that can secrete heparanase are engineered to promote infiltration and antitumor activity.
As a result, a solution against solid tumors requires a "super T cell", or other forms of ACT that may be better suited for solid tumors.
Structure and Generations of Car-T Cell
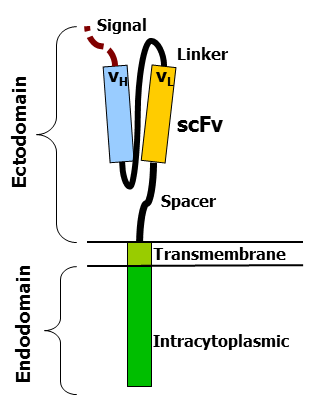
CARs are composed of three regions: the ectodomain, the transmembrane domain, and the endodomain. The ectodomain consists of three components: a signaling peptide, an antigen recognition region, and a spacer. The signal peptide directs the nascent protein into the endoplasmic reticulum. The antigen recognition region in CAR is called a single-chain variable fragment (scFv), a type of protein known as a fusion protein or chimeric protein. A scFv is a chimeric protein made up of the light and heavy chains of immunoglobins connected with a short linker peptide. The linker consists of hydrophilic residues with sections of glycine and serine in it for flexibility as well as sections of glutamate and lysine for added solubility.
The transmembrane domain is a hydrophobic alpha helix that spans the membrane. The transmembrane domain is essential for the stability of the receptor as a whole. The CD28 transmembrane domain is currently the most stable of the domains.
With the development of co-stimulatory molecules, CAR-T cells have experienced four generations of development. Many co-stimulatory molecules have been investigated, including CD28, 4-1BB (CD137), CD27, and OX40 (CD134), which have been incorporated into CARs to further enhance the therapeutic effect. First-generation CARs have a CD3ζ signaling domain to provide an activation signal. However, their expansion ability and anti-tumor efficacy remain unsatisfactory.
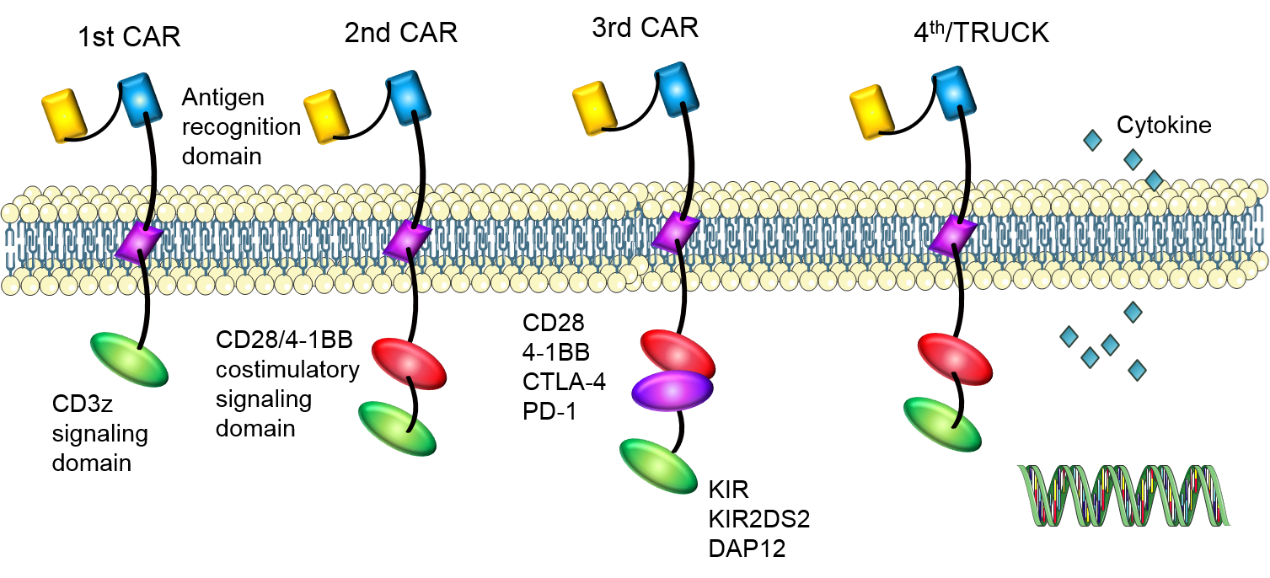
Subsequently, second-generation CARs have a costimulatory signaling domain to enhance the signal function of the CD3z signaling domain. CAR-T cells with CD28 or 4-1BB signaling domain have shown potent anti-tumor efficacy in vivo for B cell malignancies. ICOS co-stimulatory domain also has been used and CARs with ICOS tend to have enhanced survival time than CARs with CD28 or 4-1BB co-stimulatory domain.
In third-generation CARs, two co-stimulatory signaling domains are added to amplify the anti-tumor effect of second-generation CARs. CAR-T cells with CD28 and 4-1BB domains have shown enhanced functionality and increased persistence. In addition to these co-stimulatory molecules mentioned above, some other molecules are also being studied, such as CTLA-4 or PD-1, preventing the damage of inadequate T cell specificity to normal tissues. Stimulatory killer immunoglobulin-like receptor (KIR) KIR2DS2 and DNAX-activating protein of 12 kDa (DAP12) also are also used to replace CD3z and co-stimulatory molecules to enhance the proliferation and function of CAR-T cells, which can destroy immunotherapy-resistant solid tumors efficiently.
In the fourth generation CARs (TRUCKs), cytokine genes are added to CAR-T cells. CAR-T cells modified with cytokine genes can use some valid components of the tumor microenvironment to amplify anti-tumor efficacy.
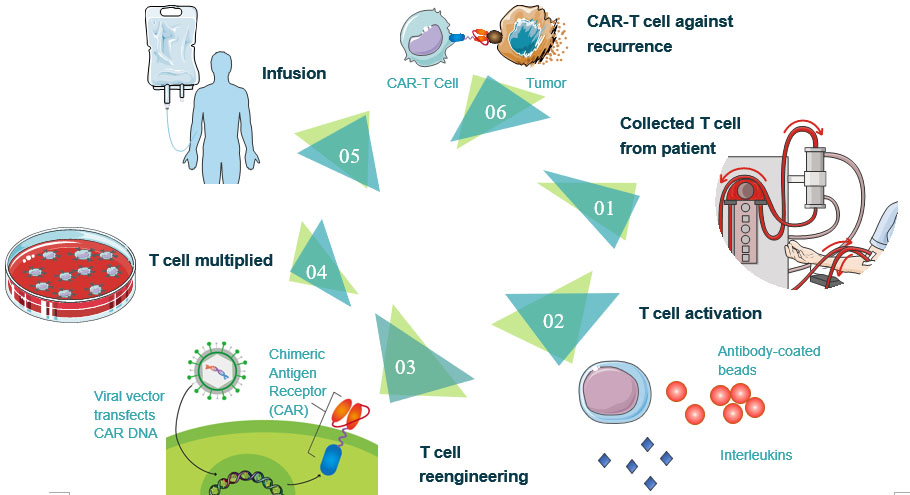
The process of CAR-T cell therapy
1. Collected T cell from the patient. T cells are collected via leukocyte apheresis. The leukocytes are removed using a blood cell separator. The patient's autologous peripheral blood mononuclear cells (PBMC) are then separated and collected from the buffy coat that formed after separation. The remaining blood is then returned to the body.
2. T cell activation. The products of leukocyte apheresis are then transferred into a cell processing center. In the cell processing center, specific T-cells are activated in a certain environment in which they can actively proliferate. The cells are activated using a type of cytokine called Interleukin 2 (IL-2) as well as anti-CD3 antibodies.
3. T cell reengineering. The T cells are sent to a laboratory or a drug manufacturing facility where they are genetically engineered by introducing DNA into them using an integrating gammaretrovirus (RV) or lentivirus (LV) vectors to produce chimeric antigen receptors (CARs) on the surface of the cells. These vectors are now very safe due to a partial deletion of the U3 region.
4. T cell multiplied. The population of genetically modified T cells is expanded by growing cells in the laboratory. When there are enough of them, these CAR-T cells are frozen and sent to the hospital or center where the patient is being treated.
5. CAR-T cells infusion. The patient undergoes lymphodepletion chemotherapy prior to the introduction of the engineered CD CAR-T cells. The depletion of the number of circulating leukocytes in the patient upregulates the number of cytokines that are produced which helps promote the expansion of the engineered CAR-T cell. CAR-T cells that have been returned to the patient's bloodstream multiply in number. These are the "attacker" cells that will recognize and attack cells that have the targeted antigen on their surface.
6. CAR-T cell against recurrence. CAR-T cells may eradicate all of the cancer cells and may remain in the body months after the infusion has been completed. The therapy has resulted in long-term remissions for some types of blood cancer.
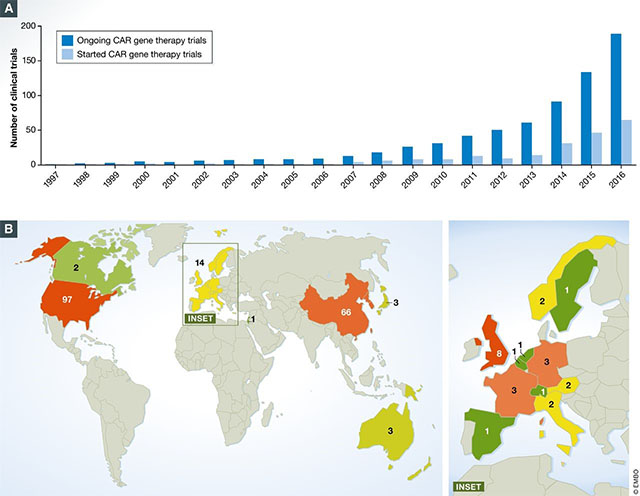
Car-T Cell Therapy Clinical Trial and Side Effect
As of August 2017, there were around 200 clinical trials happening globally involving CAR-T cells. Of those trials, around 65% were trials in which hematological malignancies were explored, and 80% of them involved CD19 CAR-T cells targeting the B-cell cancers. Studies began in 2016 to explore the viability of other antigens such as CD20.
CAR-T clinical trials have shown high remission rates of up to 94% in severe cancer types. This is particularly impressive considering that most CAR-T clinical trials recruit cancer patients that have not responded to many if no other available treatments. These results have fed the expectations of patients and investors alike, but it's important to remember that the therapy can also have downsides.
Cytokine-Release Syndrome (CRS)
Like all cancer therapies, CAR T-cell therapy can cause several worrisome, and sometimes fatal side effects.
As part of their immune-related duties, T cells release cytokines that are chemical messengers that help to stimulate and direct the immune response, causing a rapid and massive release of cytokines into the bloodstream, which can lead to dangerously high fevers and important drops in blood pressure. CRS symptoms can range from mild flu-like symptoms that include nausea, fatigue, headache, chills, and fever to more serious symptoms, such as low blood pressure, tachycardia (abnormally rapid heart rate), capillary leakage (fluid and proteins leak out of tiny blood vessels and flow into surrounding tissues, resulting in dangerously low blood pressure), cardiac arrest, cardiac arrhythmias, cardiac failure, hemophagocytic lymphohistiocytosis (life-threatening immunodeficiency)/macrophage activation syndrome (a life-threatening complication of rheumatic disease) (HLH/MAS), hypoxia (lack of oxygen reaching the tissue), renal insufficiency (poor function of the kidneys), poor lung oxygenation, and multiple organ failure. Ironically, CRS is considered an "on-target" effect of CAR-T cell therapy-that is, its presence demonstrates that active T cells are at work in the body.
Neurologic Toxicities. Common symptoms include language impairment (aphasia), confusion, delirium, involuntary muscle twitching, hallucinations, and/or unresponsiveness. Seizures have also been reported.
B-Cell Aplasia. CAR-T cell therapy targeting antigens found on the surface of B-cells does not only destroy cancerous B-cells but also normal B-cells. CD19 is also expressed on normal B-cells, which are responsible for producing antibodies that kill pathogens. These normal B-cells are also often killed by the infused CAR-T cells. To compensate for this side effect, many patients must receive immunoglobulin therapy, which provides them with the necessary antibodies to fight off infections.
Tumor Lysis Syndrome (TLS). TLS describes a group of metabolic complications that can occur due to the breakdown of dying cells - usually at the onset of toxic cancer treatments.
Anaphylaxis (Life-threatening Allergic Reaction). There is potential for a patient receiving CAR-T cell therapy to have an overwhelming immune response against the CAR itself, called "anaphylaxis". Symptoms associated with anaphylaxis include hives, facial swelling, low blood pressure, and respiratory distress.
While these are the known side effects, CAR-T cell therapy is a new treatment and doctors continue monitoring patients to uncover any long-term impact on their bodies.
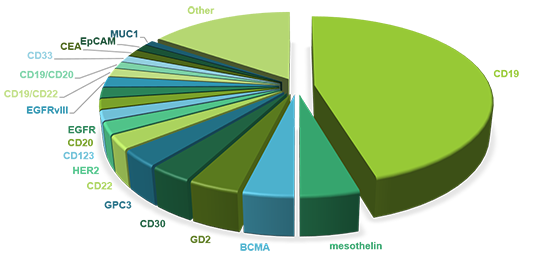
Targets of Car-T Cell Therapy
The identification of targets is a prerequisite of CAR-T cell therapy and thus happens before treatment. In order to avoid damage to healthy tissue caused by CAR-T cell therapy, the targets must be restricted to tumor cells. So far, a variety of tumor-associated antigens (TAAs) have been targeted to achieve an ideal therapeutic effect.
Table 1: CAR-T-cell targets for the treatment of hematological tumors
| Target | CAR structure | Malignancy |
| BCMA | CD3ζ and 41BB | MM |
| CD19 | CD3ζ and CD28; CD3ζ and 41BB KIR2DS2 and DAP12- |
Lymphoma; Leukemia |
| CD22 | CD3ζ and CD28 | FL; NHL; DLBCL; ALL |
| CD20 | CD3ζ; CD3ζ and 41BB- | CD20positive malignancies |
| CD138 | CD3ζ and 41BB | MM |
| CD33 | CD3ζ and 41BB | AML |
| CD123 | CD3ζ and CD28 | AML |
| CD19 CD20 |
CD3ζ and 41BB | Leukemia; Lymphoma |
| CD19 PSMA |
CD3ζ and CD28 PD-1 or CTLA4 |
Leukemias |
| FITC-CD19 Ab | CD3ζ and CD28 | CD19 positive cancers |
| Igκ | CD3ζ and CD28 | CLL |
| LeY | CD3ζ and CD28 | AML |
| ROR1 | CD3ζ and 41BB | CLL; SLL |
Table 2. CAR-T-cell targets for the treatment of solid tumors
| Target | CAR structure | Malignancy |
| Biotin | CD3ζ, CD28 and 41BB | EGFRvIII positive cancer |
| CD171 | CD3ζ and 4-1BB; CD3ζ, CD28 and 4-1BB |
Neuroblastoma |
| EGFRvIII | CD3ζ and 41BB CD3ζ and ICOS- |
Glioma |
| FAP | CD3ζ and CD28 KIR2DS2 and DAP12- |
Mesothelioma; Lung cancer |
| FR | CD3ζ and CD27 | Ovarian cancer; Breast cancer |
| Glypican-3 | CD3ζ, CD28 and 41BB | Hepatocellular carcinoma |
| HER2 | CD3ζ and CD28 | HER2 positive cancer; Sarcoma |
| HER2 MUC1 |
CD3ζ and CD28 | Breast cancer |
| HER2 IL13Rα2 |
CD3ζ and CD28 | Glioblastoma |
| IL13Rα2 | CD3ζ; CD3ζ and 41BB CD3ζ and CD28 CD3ζ, CD28 and 41BB CD3ζ, CD28 and OX40- |
Glioma |
| Mesothelin | CD3ζ; CD3ζ and CD28 CD3ζand 41BB CD3ζ and ICOS KIR2DS2 and DAP12- |
Mesothelioma; Pancreatic cancer; Non-small cell lung cancer |
| Mesothelin CD19 |
CD3ζand 41BB | Pancreatic cancer |
| MUC1 | CD3ζ and 41BB | MUC1 positive solid tumor |
| NKG2D | CD3ζ; CD3ζ and DAP10 CD3ζ and 41BB CD3ζ and CD28 |
Ovarian cancer Ewing sarcoma |
| PSMA | CD3ζ and CD28 | Prostate cancer |
| PD1 and CD19; PD1 and Mesothelin; |
CD3ζ and CD28 CD3ζ, CD28 and 41BB |
PD-L1 positive cells |
However, it is quite difficult to discover an ideal biomarker as an anti-tumor target, which highly expressed on tumor cell surface and rarely expressed on the healthy cell. In a more common situation, the chosen biomarkers express on both tumor cell and healthy ones, with a significant expression level discrimination. That is why we should pay close attention to the on-target-off-tumor cytotoxicity of CAR-T cell therapy.
The cluster of differentiation 19 (CD19) is regarded as a star biomarker in the area of CAR-T therapy. Besides CD19, other biomarkers targeting numerous different types of tumor cells are used as CAR-T targets for clinical trials. We will introduce some of the target antigens here.
CD19
The human CD19 antigen is a 95 kD transmembrane glycoprotein belonging to the immunoglobulin (Ig) superfamily. CD19 is classified as a type I transmembrane protein, with a single transmembrane domain, a cytoplasmic C-terminus, and extracellular N-terminus. CD19 is critically involved in establishing intrinsic B cell signaling thresholds through modulating both B cell receptor (BCR)-dependent and independent signaling. It plays roles in the antigen-independent development as well as the immunoglobulin-induced activation of B cells. CD19 is thus critical for the body to mount an optimal immune response. On B cells, CD19 associates with CD21, CD81, and CD225, forming a signal transduction complex. Expression of CD19 is found in the majority of B cell-derived malignancies, as well as on follicular dendritic cells.
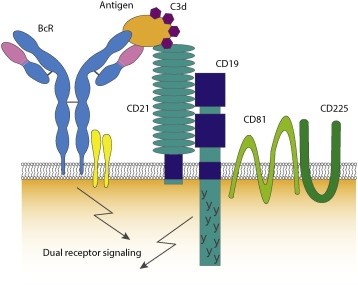
The CD19 antigen plays an important role in clinical oncology. CD19-targeted CARs to treat B cell cancers are up to 70%–90% response rate in acute and chronic leukemias. Although the great success with CD19 specific CARs, CD19 escape variants have been confirmed after therapy and responding patients with subsequent target loss have a recurrence of the disease. To overcome such antigen target issues about escape variants, one method is to investigate other tumor antigen targets, such as CD22, CD20, CD138, CD33, CD123, and so on.
CD20
CD20 was discovered in 1980 as the first specific B-cell marker. It is a non-glycosylated tetraspanin of the membrane spanning 4-A family, with two extracellular loops. CD20 is expressed on the surface of all B-cells beginning at the pro-B phase (CD45R+, CD117+) and progressively increasing in concentration until maturity. It is suspected that it acts as a calcium channel in the cell membrane and is thought to modulate calcium release arising from the BCR.
Importantly, as well as being expressed on normal B-cells, CD20 was also found to be expressed on the surface of malignant B-cells. It is highly expressed, with approximately 100,000 CD20 molecules expressed on the surface of most malignant cells.
BCMA
B-cell maturation antigen (BCMA or BCM), also known as tumor necrosis factor receptor superfamily member 17 (TNFRSF17), is predominantly expressed on terminally differentiated B cells and, upon binHERding to its ligands B cell activator of the TNF family (BAFF) and a proliferation-inducing ligand (APRIL), delivers pro-survival cell signals. Thus, BCMA is most known for its functional activity in mediating the survival of plasma cells that maintain long-term humoral immunity. The expression of BCMA has been also linked to a number of cancers, autoimmune disorders, and infectious diseases that suggest additional roles for BCMA activity.
BCMA is expressed on a number of hematologic malignancies, such as Hodgkin’s and non-Hodgkin’s lymphomas. BCMA could maintain the viability and proliferation of malignant cells. BCMA expression is also found in both primary tumor cells and cell lines of multiple myeloma. A critical role for BCMA in protecting myeloma cells from apoptosis. BCMA-specific antagonists may have significant clinical benefit for BCMA-expressing cancers.
HER2
Receptor tyrosine-protein kinase erbB-2, also known as CD340 (cluster of differentiation 340), proto-oncogene Neu, Erbb2 (rodent), or ERBB2 (human), is also frequently called HER2 (from human epidermal growth factor receptor 2) or HER2/neu.
HER2 is a non-ligand-binding member of this family and exerts its activity through heterodimerisation with other EGFR family members. HER2 functional activation promotes oncogenesis. More recently, somatic HER2 gene mutations have been detected in a range of human cancer types.
Amplification, also known as the over-expression of the ERBB2 gene, occurs in approximately 15-30% of breast cancers. It is strongly associated with increased disease recurrence and a poor prognosis. Over-expression is also known to occur in ovarian, stomach, adenocarcinoma of the lung and aggressive forms of uterine cancer, such as uterine serous endometrial carcinoma.
To overcome target antigen escaping variants, except investigating other tumor antigen targets, the other method is the concepts of double CARs or dual receptors within one T cell, switchable CARs mentioned above also can be used here to prevent the development of antigen escape variants.
Case Study
Case 1: Good Z, Spiegel JY, Sahaf B, et al. Post-infusion CAR TReg cells identify patients resistant to CD19-CAR therapy. Nat Med. 2022 Sep;28(9):1860-1871. doi: 10.1038/s41591-022-01960-7. Epub 2022 Sep 12. PMID: 36097223; PMCID: PMC10917089.
Approximately 60% of patients with large B cell lymphoma treated with chimeric antigen receptor (CAR) T cell therapies targeting CD19 experience disease progression, and neurotoxicity remains a challenge. Biomarkers associated with resistance and toxicity are limited. In this study, single-cell proteomic profiling of circulating CAR T cells in 32 patients treated with CD19-CAR identified that CD4+Helios+ CAR T cells on day 7 after infusion are associated with progressive disease and less severe neurotoxicity. These data credential CAR TReg cell expansion as a novel biomarker of response and toxicity after CAR T cell therapy and raise the prospect that this subset may regulate CAR T cell responses in humans.
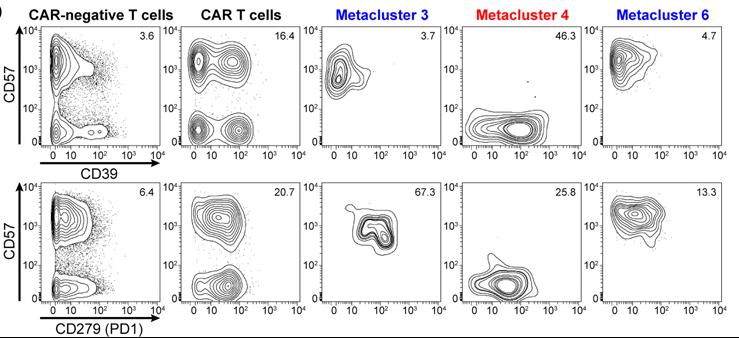
Fig1. Protein expression in CyTOF metaclusters of circulating CAR T cells on day 7. Contour plots showing expression of exhaustion markers CD39 and CD279 (PD1) against senescence marker CD57 in CAR− and CAR+ T cells, as well as in CAR T cell metaclusters 3, 4, and 6, for patient 004.
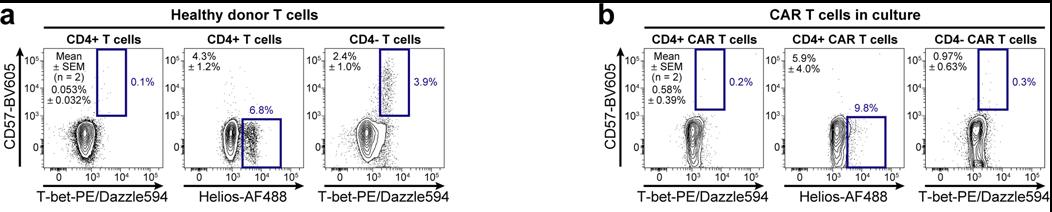
Fig2. Identified populations in healthy donors and CD19-CD28ζ CAR-transduced T cells. a-b, Contour plots show three identified populations among T cells from a healthy donor (a) and among CD19-CD28ζ CAR-transduced T cells generated in the lab (b). Population statistics for two donors are shown as mean ± SEM on each plot.
Case 2: Mailankody S, Devlin SM, Landa J, et al. GPRC5D-Targeted CAR T Cells for Myeloma. N Engl J Med. 2022 Sep 29;387(13):1196-1206. doi: 10.1056/NEJMoa2209900. PMID: 36170501; PMCID: PMC10309537.
B-cell maturation antigen (BCMA)-directed chimeric antigen receptor (CAR) T-cell therapies have generated responses in patients with advanced myeloma, but relapses are common. G protein-coupled receptor, class C, group 5, member D (GPRC5D) has been identified as an immunotherapeutic target in multiple myeloma. Preclinical studies have shown the efficacy of GPRC5D-targeted CAR T cells, including activity in a BCMA antigen escape model.
In this phase 1 dose-escalation study, we administered a GPRC5D-targeted CAR T-cell therapy (MCARH109) at four dose levels to patients with heavily pretreated multiple myeloma, including patients with relapse after BCMA CAR T-cell therapy.
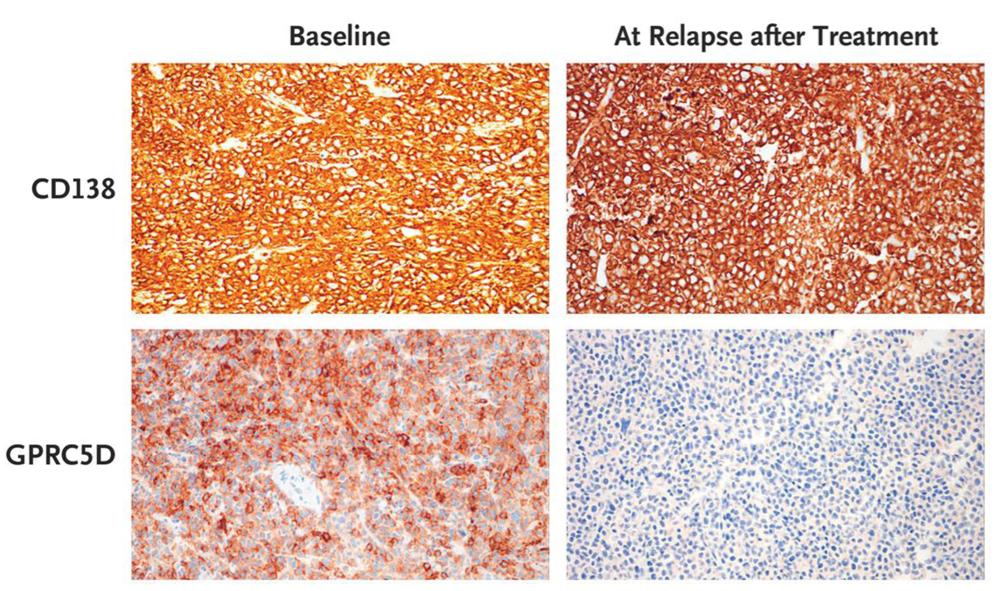
Fig1. Loss of GPRC5D on Immunohistochemical Analysis at Relapse after MCARH109 Infusion. GPRC5D and CD138 immunohistochemical analysis of plasmacytoma tissue samples from Patient 15 at baseline and at relapse after MCARH109 infusion (dose level, 150×106 CAR T cells) shows loss of GPRC5D expression at the time of relapse. The patient had a best response of stringent complete response.
Case 3: Jain N, Zhao Z, Feucht J, et al. TET2 guards against unchecked BATF3-induced CAR T cell expansion. Nature. 2023 Mar;615(7951):315-322. doi: 10.1038/s41586-022-05692-z. Epub 2023 Feb 8. PMID: 36755094; PMCID: PMC10511001.
As T cell differentiation and functional states are associated with distinct epigenetic profiles, the authors hypothesized that epigenetic programming may provide a means to improve CAR T cell performance. Targeting the gene that encodes the epigenetic regulator ten-eleven translocation 2 (TET2) presents an interesting opportunity as its loss may enhance T cell memory, albeit not cause malignancy. Here they show that disruption of TET2 enhances T cell-mediated tumour rejection in leukaemia and prostate cancer models. However, loss of TET2 also enables antigen-independent CAR T cell clonal expansions that may eventually result in prominent systemic tissue infiltration.

Fig1. Effect of TET2 editing on CAR T cell accumulation in a prostate cancer model. b, CAR T cell counts in the peripheral blood 30 days post infusion of T cells. Bars show median values. c, Mice with the top 4 CAR T cell peripheral counts at day 30 across both TET2 targeting gRNA (g1, n=2. g2, n=2) were euthanized at day 45 along with 5 scrambled gRNA treated PSMA28z+41BBL mice and their splenic CAR T cell numbers were quantified. p values were determined by two-sided Mann-Whitney
Related Resource
 Download PDF
Download PDF