
Autophagy
Creative BioMart Autophagy Product List
Immunology Background
Background
Autophagy, derived from the Greek words "auto" meaning self and "phagy" meaning eating, represents a fundamental cellular process whereby cells degrade and recycle their own components. This encompasses a range of substances, from misfolded proteins to entire cell organelles. This self-digestive mechanism plays a pivotal role in maintaining cellular homeostasis, responding to stress, and influencing a multitude of physiological and pathological processes.
Definition
Autophagy is a lysosome-dependent degradation pathway whereby cellular components, including damaged organelles, misfolded proteins, and invading pathogens, are engulfed by autophagosomes and delivered to lysosomes for degradation and recycling. This process enables cells to withstand stressful conditions, such as nutrient deprivation, and to maintain cellular quality control by eliminating defective components.
Autophagy is a process that occurs in a wide variety of eukaryotic organisms, including plants, multicellular animals, slime molds, and yeasts. The process is essential for maintaining equilibrium between the synthesis of new cellular components and the degradation of existing ones. A mitochondrion within a liver cell, for instance, has a lifespan of approximately ten days before it is degraded by autophagy (mitophagy, in part via the omegasome) and its components are reused to construct other structures (salvage pathway).
Classification
Autophagy can be classified into three main types based on the method of cargo delivery to lysosomes:
Macroautophagy
Macroautophagy represents the primary mechanism of autophagy, facilitating the degradation of cellular organelles and proteins. In this process, a portion of the endoplasmic reticulum (ER) membrane encloses the structures designated for degradation until the organelle to be degraded is completely enclosed by a double bio membrane. Subsequently, the autophagosome is transported along the actin cytoskeleton until it ultimately binds to a lysosome via membrane fusion, whereupon its contents are degraded by lysosomal acid hydrolases.
Microautophagy
In this type, the lysosomal membrane directly engulfs small portions of cytoplasm through invagination, which are then degraded.
Chaperone-mediated autophagy (CMA)
In chaperone-mediated autophagy (CMA), proteins bearing a KFERQ-like motif are identified by the chaperone or heat shock protein Hsc70 and transported to the lysosome as a protein complex. There, they are transported into the lysosome by the lysosomal membrane-associated protein 2a (LAMP-2A) and degraded. The complex is then internalized into the lysosome via receptor-mediated endocytosis, with Hsc70 acting as a chaperone to facilitate the unfolding of the imported proteins. It has been demonstrated that altered chaperone-mediated autophagy plays a role in the development of Parkinson's disease.
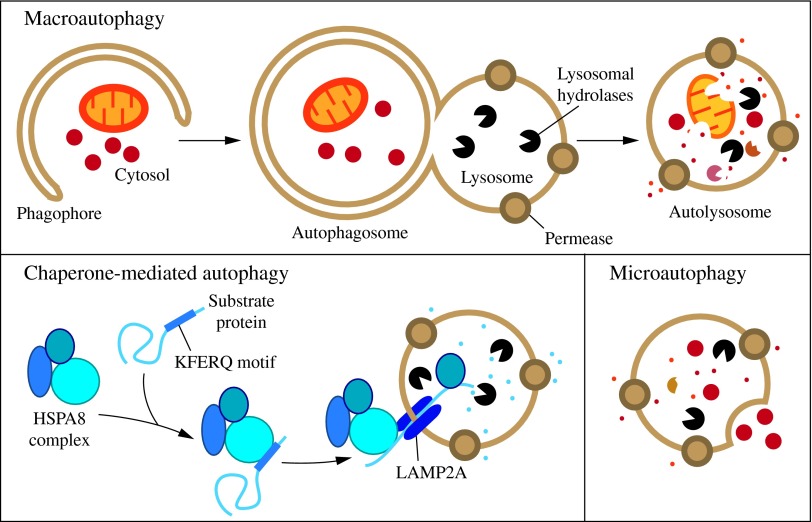 Fig.1: Three types of autophagy in mammalian cells (Parzych and Klionsky, 2014).
Fig.1: Three types of autophagy in mammalian cells (Parzych and Klionsky, 2014).Mechanism of Autophagy
The autophagy process can be divided into several stages:
Initiation: The process commences with the formation of the phagophore, a small, double-membrane structure. This process is regulated by the Unc-51 Like Autophagy Activating Kinase 1 (ULK1) complex, which is activated in response to stress or nutrient deprivation.
Nucleation: The phagophore expands and forms a pre-autophagosomal structure with the help of the Beclin-1 complex, which includes Class III phosphatidylinositol 3-kinase (PI3K).
Expansion and Maturation: The phagophore elongates and engulfs the cytoplasmic material, forming an autophagosome. The phagophore elongates and engulfs the cytoplasmic material, forming an autophagosome. This step involves two ubiquitin-like conjugation systems: the ATG12-ATG5-ATG16L complex and the LC3-II conjugation system.
Fusion with Lysosome: The mature autophagosome fuses with the lysosome, forming an autolysosome. This process is facilitated by the involvement of SNARE proteins and other fusion machinery.
Degradation: The cargo within the autolysosome is degraded by lysosomal hydrolases, and the resulting macromolecules are released back into the cytoplasm for reuse.
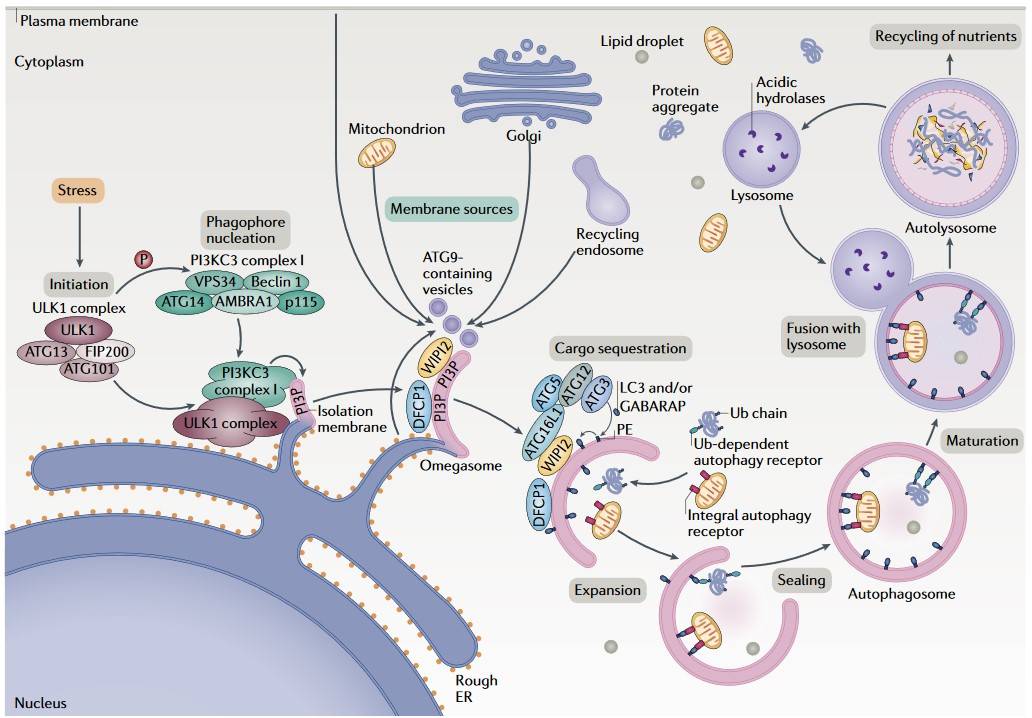 Fig. 2: Overview of the autophagy process (Dikic and Zvulun, 2018).
Fig. 2: Overview of the autophagy process (Dikic and Zvulun, 2018).Physiological Role of Autophagy
Autophagy is significant for several vital cellular functions, including:
Cellular Homeostasis
It maintains cellular homeostasis by degrading and recycling damaged organelles and proteins, thereby preventing the accumulation of toxic materials. Neurons, as post-mitotic cells, are largely dependent on autophagy to maintain cellular homeostasis by removing damaged and/or old organelles and misfolded or aggregated proteins.
Response to Stress
Autophagy is involved in the degradation and remodeling of proteins and lipids and in the provision of amino acids when food intake is reduced or (partially) withheld, as is the case during fasting. Fasting induces autophagy, which occurs to a lesser extent in all cells, but is enhanced during metabolic, genotoxic, infectious, and hypoxic stress.
Development and Differentiation
Autophagy is involved in cellular differentiation and development, as seen in processes such as erythropoiesis and adipogenesis, and is essential for the removal of mitochondria from immature erythrocytes, a process known as mitophagy. This mitochondrial clearance is critical because mature erythrocytes lack organelles to optimize their oxygen-carrying capacity. Disruption of autophagy during erythropoiesis can lead to defective red blood cell maturation and contribute to anemia.
Immune Defense
Autophagy also enables the intracellular degradation of viruses, bacteria and foreign proteins that have invaded the cell. In addition to degradation, it also serves the immune response, as antigen presentation occurs subsequently via MHC I and II. Several intracellular bacterial pathogens have developed resistance mechanisms against autophagic degradation.
Quality Control
Autophagy ensures the quality control of proteins and organelles, thus preventing diseases associated with the accumulation of damaged components.
Causes of Autophagy
Autophagy can be triggered by several factors, including nutrient deprivation, hypoxia, oxidative stress, pathogen invasion, and protein aggregation. Starvation activates autophagy to provide essential nutrients and energy by degrading cellular components. Low oxygen levels can induce autophagy as a survival mechanism. The accumulation of reactive oxygen species (ROS) can also activate autophagy to remove damaged mitochondria (mitophagy) and reduce oxidative damage. In addition, bacterial or viral infection can trigger autophagy as part of the immune response to degrade the pathogens. And the presence of misfolded or aggregated proteins can stimulate autophagy to prevent cellular toxicity.
Regulation of Autophagy
The mechanistic target of rapamycin (mTOR) is a key negative regulator of autophagy. This serine/threonine kinase acts as a central sensor of cellular nutrient, energy and oxygen levels and integrates these signals to regulate cell growth, proliferation and survival. Under nutrient-rich conditions, mTORC1 (mammalian target of rapamycin complex 1) is activated and inhibits the activity of the ULK1 complex by phosphorylating ULK1 and ATG13. The nucleation step of autophagy is inhibited by the phosphorylation of ATG14, AMBRA1 and NRBF2 in the PI3KC3 complex I. Phosphorylation of p300 and WIPI2 by mTORC1 inhibits VSP34 activity/LC3 lipidation and the recruitment of phosphatidylinositol phosphates along with the LC3 conjugation system for the autophagosome elongation. Finally, mTORC1 negatively regulates the fusion of the autophagosome with the lysosome through the phosphorylation of UVRAG and Pacer, which are important for the lipid kinase activity of PI3KC3 complex II and the recruitment of the HOPS tethering complex. Conversely, under conditions of nutrient deprivation or starvation, mTOR activity is suppressed, often through upstream signals such as low amino acid levels, decreased ATP, and increased AMP/ATP ratio, leading to the activation of AMP-activated protein kinase (AMPK).
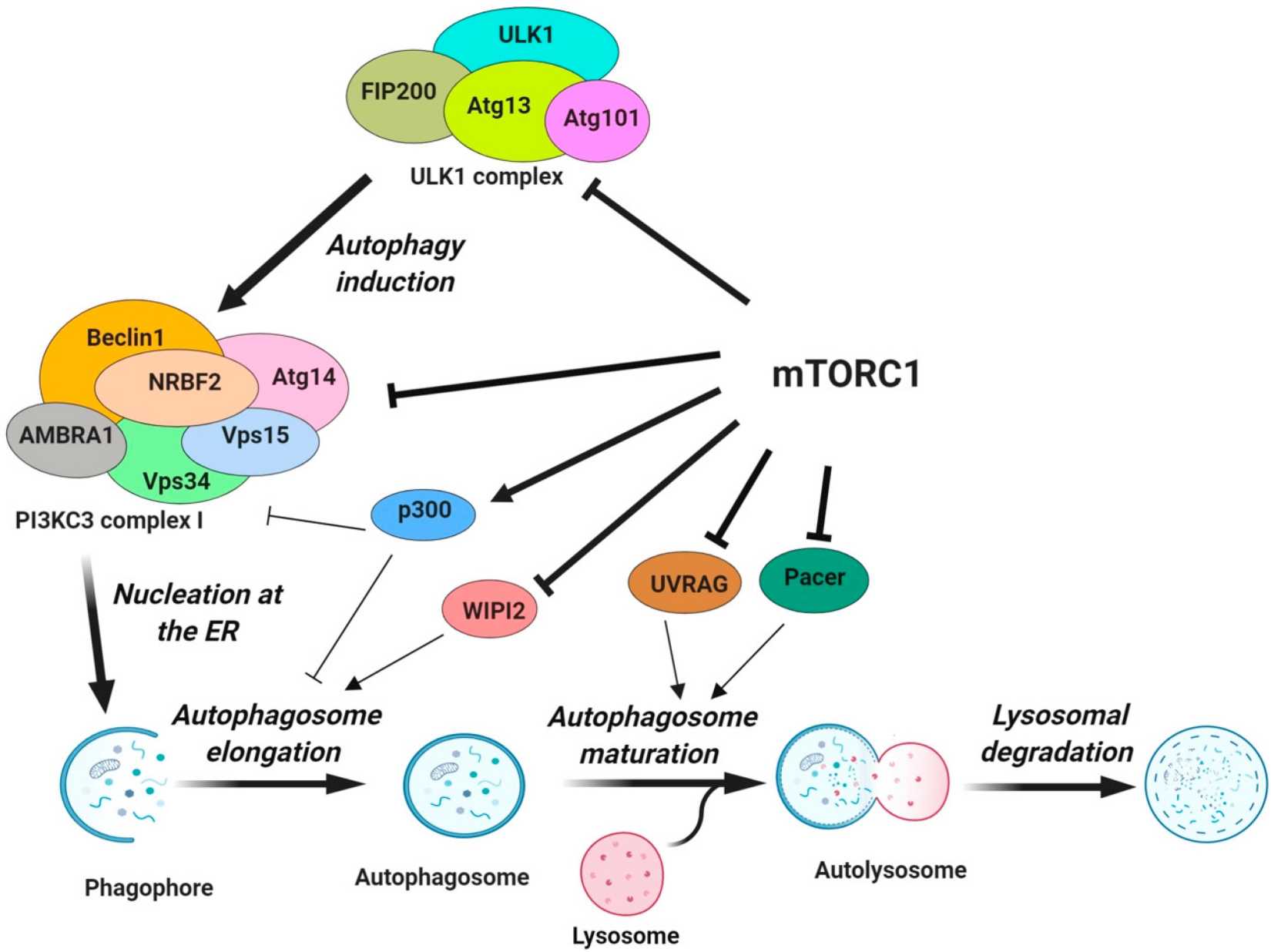 Fig. 3: Regulation of various steps of autophagy by mTORC1. mTORC1 inhibits the activity of the ULK1 complex by phosphorylating ULK1 and ATG13 (Dossou and Basu, 2019).
Fig. 3: Regulation of various steps of autophagy by mTORC1. mTORC1 inhibits the activity of the ULK1 complex by phosphorylating ULK1 and ATG13 (Dossou and Basu, 2019).AMPK Pathway
AMP-activated protein kinase (AMPK) is an evolutionarily conserved serine/threonine protein kinase and energy sensor that controls autophagy through three levels of regulation. First, AMPK represses mTOR activity by phosphorylating TSC2 and RPTOR. MPK directly phosphorylates the mTORC1 component RPTOR. This phosphorylation induces YWHA/14-3-3 binding to RPTOR, which prevents RPTOR from binding to mTOR and mTOR substrates, leading to suppression of mTORC1 activity. Alternatively, AMPK phosphorylates the upstream regulator of mTOR, TSC, which promotes the GTPase-activating function of the TSC1-TSC2 complex, resulting in the conversion of RHEB to an inactive RHEB-GDP state, thereby reducing mTOR activity. Second, AMPK directly phosphorylates and activates proteins involved in autophagy, including ULK1, BECN1, PIK3C3/VPS34, and ATG9A.
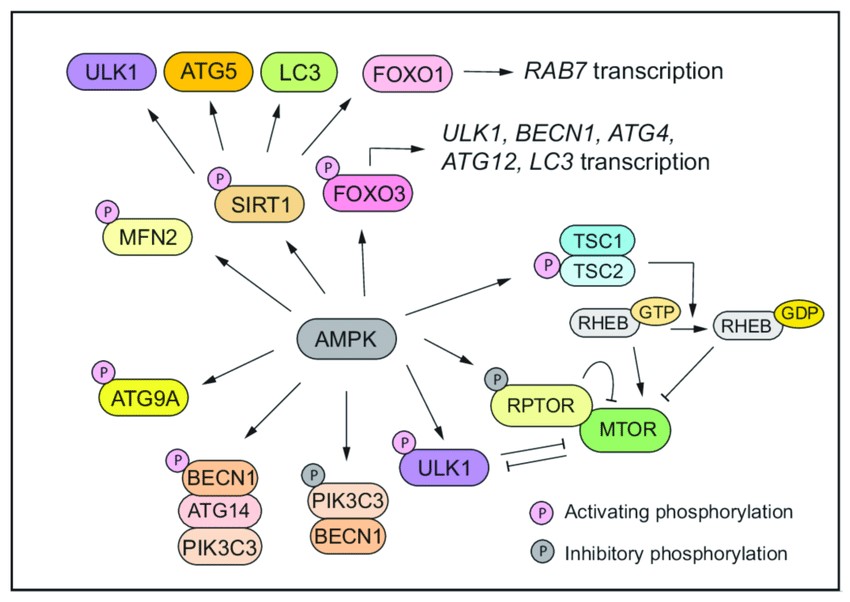 Fig. 4: AMPK drives autophagy through three layers of regulation (Lei et al., 2022).
Fig. 4: AMPK drives autophagy through three layers of regulation (Lei et al., 2022).Apoptosis, Necrosis, and Autophagy
Autophagy, apoptosis, and necroptosis are separate processes that are essential for maintaining cellular balance, development, and disease progression. Apoptosis is a form of caspase-driven programmed cell death characterized by chromatin condensation, nuclear fragmentation, and membrane blebbing. Necrosis, on the other hand, is considered to be unregulated, random cell death caused by non-specific or non-physiological stress factors, characterized by swelling of cellular organelles, rupture of the plasma membrane, and subsequent inflammatory responses due to the release of intracellular contents. Autophagy is primarily a survival mechanism but can also lead to autophagic cell death. Although the molecular mechanisms of these types of cell death are distinct, they overlap. Different signaling pathways independently regulate these forms of cell death; however, they are interconnected, can be activated simultaneously, and can function in parallel in cells under stress.
 Fig. 5: Survival superiority among the different types of cell death (Chen, Kang, and Fu, 2018).
Fig. 5: Survival superiority among the different types of cell death (Chen, Kang, and Fu, 2018).Relationship with Diseases
Autophagy can act as a tumor suppressor by preventing the accumulation of damaged organelles and proteins. However, in established tumors, autophagy can promote cancer cell survival under stressful conditions such as hypoxia and nutrient deprivation. Therefore, autophagy modulation is being explored as a therapeutic strategy in cancer treatment.
Dysregulated autophagy has been implicated in neurodegenerative diseases such as Alzheimer's, Parkinson's and Huntington's disease. The accumulation of protein aggregates and damaged organelles due to defective autophagy contributes to neuronal dysfunction and degeneration.
Infectious Diseases
Autophagy plays a critical role in the immune response to infection by degrading intracellular pathogens. However, some pathogens have evolved mechanisms to evade or exploit autophagy for replication.
Autophagy is involved in the maintenance of cardiac homeostasis. Dysregulation of autophagy may contribute to cardiac diseases such as ischemia/reperfusion injury, heart failure and hypertrophy.
Autophagy is essential for lipid metabolism and glucose homeostasis. Dysregulated autophagy is associated with metabolic disorders like obesity, diabetes, and fatty liver disease.
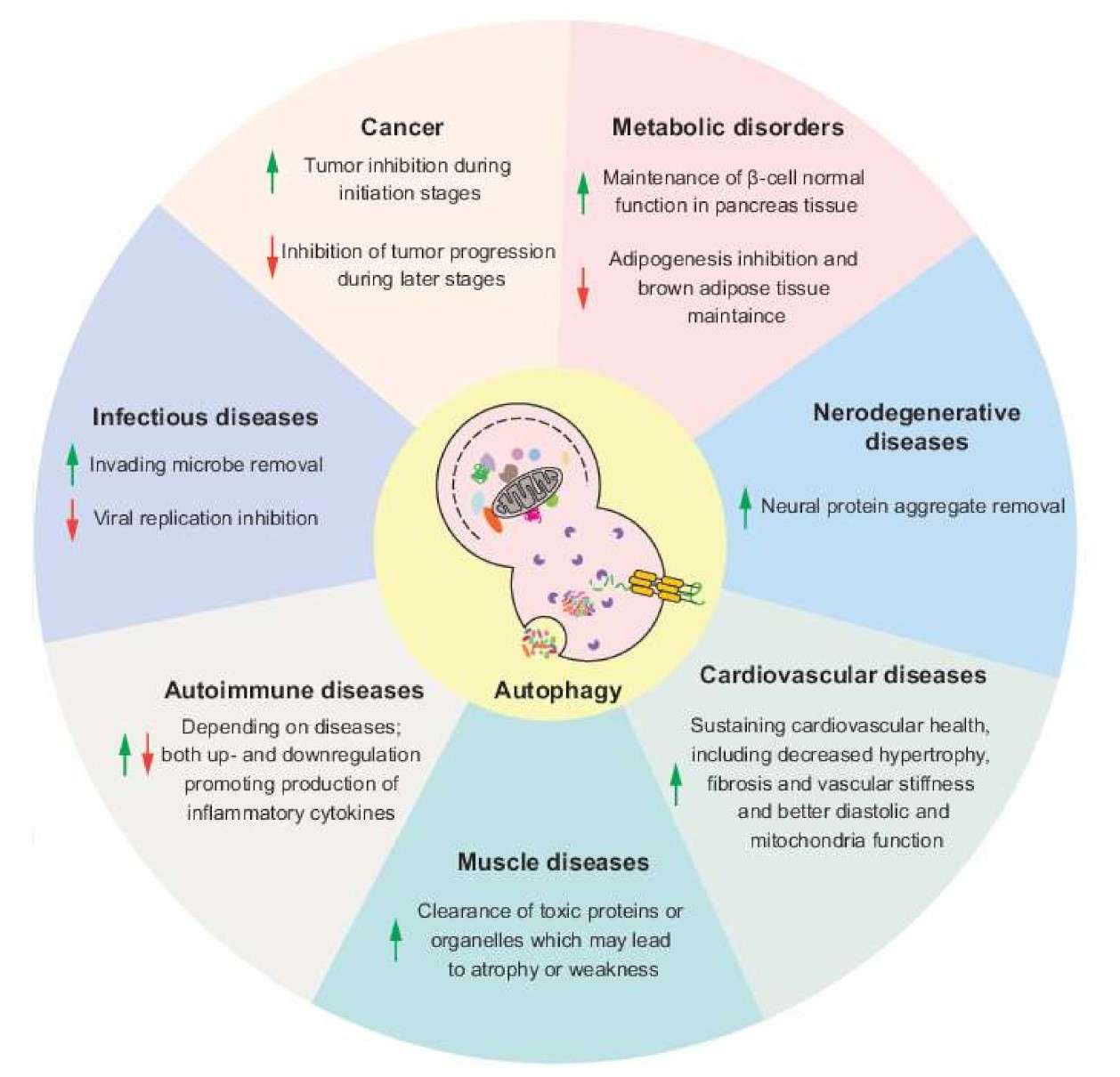 Fig. 6: Potential effects of autophagy modulation in different human diseases. Arrows indicate the upregulation or downregulation of autophagy (Lei and Klionsky, 2021).
Fig. 6: Potential effects of autophagy modulation in different human diseases. Arrows indicate the upregulation or downregulation of autophagy (Lei and Klionsky, 2021).Case Study
Case 1: Caramés, B.; et al. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015 Jun;67(6):1568-76.
Osteoarthritis (OA) is a degenerative joint disease in which the tissues in the joint break down over time. Aging is a major risk factor for OA. Recently, autophagy defects have been implicated in OA articular cartilage. To investigate the relationship between autophagy defects and cartilage damage during joint aging, a GFP-LC3 transgenic mouse model was generated. GFP-LC3 was quantified by confocal microscopy as a measure of autophagy activation. Expression of the autophagy proteins Atg5 and LC3 was analyzed by immunohistochemistry. The results showed that the expression of Atg5 and LC3 decreased with age, followed by a decrease in cartilage cellularity and an increase in the apoptosis marker PARP p85. Cartilage structural damage progressed in an age-dependent manner after autophagy changes.
Immunohistochemistry was performed for Atg5, an autophagy regulator, and LC3, an autophagy effector, in knee cartilage of mice at 6, 18, 24 and 28 months of age. The expression of Atg5 was already significantly (p<0.01) reduced in 18-month-old mice, with only small and non-significant further decreases at 24 and 28 months. LC3 was significantly reduced at 24 and 28 months of age. These results indicate that key autophagy proteins are reduced with aging.
Considering the findings presented in this study, it is reasonable to suggest that autophagy defects may play a role in the pathogenesis of osteoarthritis (OA).
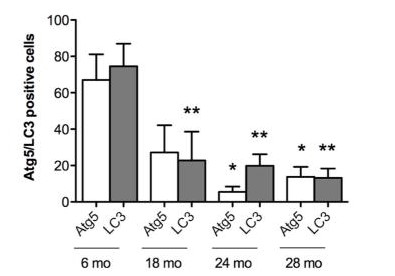 Fig. 7: Aging-associated changes in expression of autophagy proteins. Quantitative analysis of Atg5 and LC3-positive cells. Total cell number in three fields and Atg5 and LC3-positive cells were counted, and the percentage of positive cells was calculated. Results show a significant decrease in Atg5 and LC3. Values are mean ± SEM. * = P < 0.01; ** = P < 0.01 versus 6 months old mouse knee joints.
Fig. 7: Aging-associated changes in expression of autophagy proteins. Quantitative analysis of Atg5 and LC3-positive cells. Total cell number in three fields and Atg5 and LC3-positive cells were counted, and the percentage of positive cells was calculated. Results show a significant decrease in Atg5 and LC3. Values are mean ± SEM. * = P < 0.01; ** = P < 0.01 versus 6 months old mouse knee joints.Case 2: Qu, X.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003 Dec;112(12):1809-20.
Beclin-1 is a protein encoded by the human beclin-1 gene. Beclin-1 is a mammalian ortholog of the yeast autophagy-related gene 6 (Atg6) and BEC-1 in the nematode C. elegans nematode. This protein interacts with either BCL-2 or PI3k class III, playing a critical role in the regulation of both autophagy and cell death. In 40-75% of sporadic human breast, ovarian and prostate cancers, the beclin 1 autophagy gene is monoallelic deletion. Therefore, the researchers used a targeted mutant mouse model to test the hypothesis that beclin 1 monoallelic deletion promotes tumorigenesis. Their results showed that heterozygous disruption of beclin 1 results in increased cellular proliferation and decreased autophagy in vivo.
Approximately 100 beclin 1+/– and 100 beclin 1+/+ control littermates were sacrificed at 13-18 months of age and all major organs were examined macroscopically and microscopically. beclin 1+/– mice were significantly more likely to have a malignancy detected on gross examination.
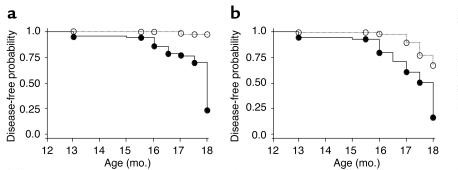 Fig. 8: Increase in the frequency of spontaneous malignancies. Kaplan-Meier plot of time to development of macroscopic malignancy (a) and any malignancy (b) in beclin 1+/– (solid lines, filled circles) versus beclin 1+/+ (dotted lines, open circles) mice (P < 0.0001, log-rank test).
Fig. 8: Increase in the frequency of spontaneous malignancies. Kaplan-Meier plot of time to development of macroscopic malignancy (a) and any malignancy (b) in beclin 1+/– (solid lines, filled circles) versus beclin 1+/+ (dotted lines, open circles) mice (P < 0.0001, log-rank test).References
- Caramés, Beatriz, et al. "The Relationship of Autophagy Defects and Cartilage Damage during Joint Aging in a Mouse Model." Arthritis & Rheumatology (Hoboken, N.J.), vol. 67, no. 6, June 2015, pp. 1568–76. PubMed Central, https://doi.org/10.1002/art.39073.
- Chen, Qi, et al. "The Independence of and Associations among Apoptosis, Autophagy, and Necrosis." Signal Transduction and Targeted Therapy, vol. 3, no. 1, July 2018, pp. 1–11. www.nature.com, https://doi.org/10.1038/s41392-018-0018-5.
- Dossou, Akpedje S., and Alakananda Basu. "The Emerging Roles of mTORC1 in Macromanaging Autophagy." Cancers, vol. 11, no. 10, Oct. 2019, p. 1422. www.mdpi.com, https://doi.org/10.3390/cancers11101422.
- Dikic, Ivan, and Zvulun Elazar. "Mechanism and Medical Implications of Mammalian Autophagy." Nature Reviews Molecular Cell Biology, vol. 19, no. 6, June 2018, pp. 349–64. www.nature.com, https://doi.org/10.1038/s41580-018-0003-4.
- Lei, Yuchen, and Daniel J. Klionsky. "The Emerging Roles of Autophagy in Human Diseases." Biomedicines, vol. 9, no. 11, Nov. 2021, p. 1651. www.mdpi.com, https://doi.org/10.3390/biomedicines9111651.
- Lei, Yuchen, et al. "How Cells Deal with the Fluctuating Environment: Autophagy Regulation under Stress in Yeast and Mammalian Systems." Antioxidants, vol. 11, no. 2, Feb. 2022, p. 304. www.mdpi.com, https://doi.org/10.3390/antiox11020304.
- Parzych, Katherine R., and Daniel J. Klionsky. "An Overview of Autophagy: Morphology, Mechanism, and Regulation." Antioxidants & Redox Signaling, vol. 20, no. 3, Jan. 2014, pp. 460–73. PubMed Central, https://doi.org/10.1089/ars.2013.5371.
- Qu, Xueping, et al. "Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene." Journal of Clinical Investigation, vol. 112, no. 12, Dec. 2003, pp. 1809–20. PubMed Central, https://doi.org/10.1172/JCI200320039.