



BCL2 Gene Family
🧪 BCL10-1016H
Source: Sf9 Cells
Species: Human
Tag: Non
Conjugation:
Protein Length:

🧪 Bid-4029M
Source: E.coli
Species: Mouse
Tag: GST&His
Conjugation:
Protein Length: Met1-Asp195
🧪 BCL2-26734TH
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-451 a.a.
🧪 BCL2L11-26430TH
Source: Wheat Germ
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-112 a.a.
🧪 BCL2L1-6872H
Source: E. coli
Species: Human
Tag: His
Conjugation:
Protein Length: 1-212 aa

🧪 BCL2A1-10178H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-175a.a.
🧪 BCL2L10-10179H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 1-172a.a.
🧪 BCL2L14-10182H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 1-327a.a.
🧪 BCL2L2-10183H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 1-171a.a.
🧪 BCL6-10185H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 183-532a.a.
🧪 BCL6B-10186H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 1-352a.a.
🧪 BCL7C-10189H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-217a.a.
🧪 BCL9-10190H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 8-340a.a.
🧪 BCL2L1-10191H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 1-233 aa
🧪 MCL1-779H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 1-350aa
SubCategories
BCL2 Family—Product Overview
The BCL2 (B-cell lymphoma 2) protein family plays a critical role in regulating apoptosis, the process of programmed cell death that is essential for maintaining cellular homeostasis. This family consists of both pro-apoptotic and anti-apoptotic proteins that form a complex regulatory network that determines cell fate. Due to their involvement in various diseases, particularly cancer, BCL2 family proteins are being widely investigated for their therapeutic potential. Creative BioMart offers a comprehensive range of recombinant BCL2 family proteins and related products to support research in apoptosis, oncology and drug development.
Jump to Section
Product Families
-
Anti-apoptotic Proteins
-
Pro-apoptotic Pore-Formers
-
Pro-apoptotic BH3-only Proteins
-
BCL2 Family-Related Proteins
Background
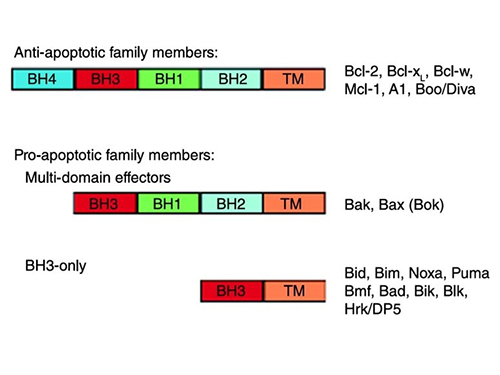
The BCL2 family was first identified through its association with follicular lymphoma, where the BCL2 gene was found to be translocated to the immunoglobulin heavy chain locus, leading to its overexpression and inhibition of apoptosis. The family is divided into three functional groups: anti-apoptotic proteins (e.g. BCL2, BCL-xL, MCL1), which promote cell survival; pro-apoptotic effectors (e.g., BAX, BAK, BOK) that induce mitochondrial outer membrane permeabilization (MOMP); and BH3-only proteins (e.g., BOD, BID, BIM) that regulate apoptosis initiation by antagonizing anti-apoptotic members or directly activating Bax/Bak.
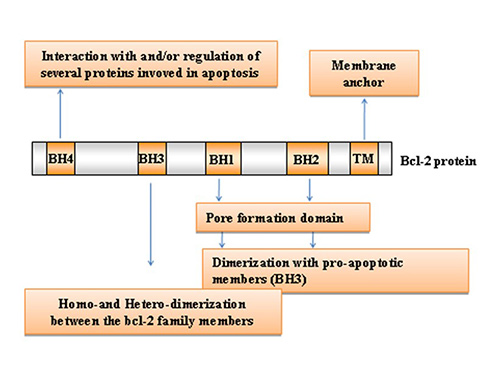
BCL2 family proteins share conserved BCL2 homology (BH) domains (BH1-4), with anti-apoptotic members typically containing all four, whereas pro-apoptotic proteins may lack one or more. The BH3 domain is particularly critical because it mediates interactions between family members that dictate survival or death signals. In addition, most members possess a C-terminal transmembrane domain that anchors them to mitochondrial, endoplasmic reticulum, or nuclear membranes, where they regulate apoptosis.
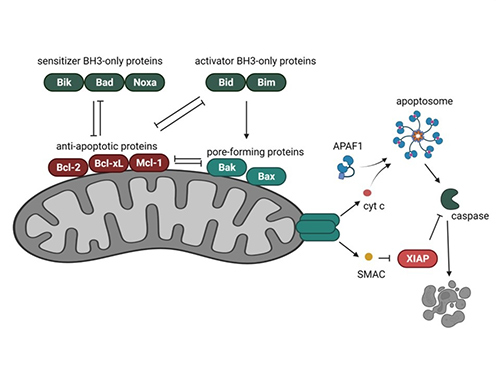
The BCL2 family regulates apoptosis through complex interactions between pro- and anti-apoptotic members. Anti-apoptotic proteins prevent mitochondrial outer membrane permeabilization (MOMP), while pro-apoptotic members promote cytochrome c release, initiating caspase activation and cell death. This balance determines cell survival, making BCL2 family proteins central to cell fate decisions.
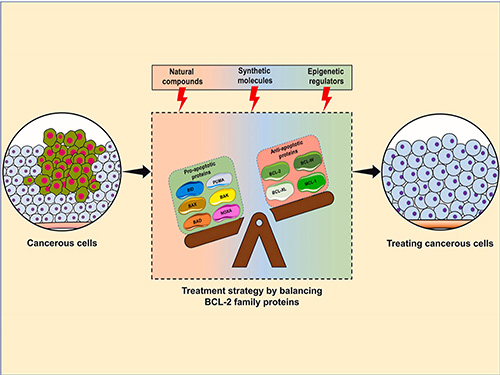
Dysregulation of the BCL2 family of proteins is a hallmark of cancer. Overexpression of anti-apoptotic proteins such as BCL2 and MCL1 allows malignant cells to evade apoptosis, leading to tumor progression and resistance to therapy. As a result, BCL2 inhibitors have been developed to selectively target these proteins, offering promising treatment strategies for hematological malignancies and solid tumors.
Applications

Cancer Research
The BCL2 family plays a critical role in regulating apoptosis, and its dysregulation is a hallmark of many cancers. Overexpression of anti-apoptotic proteins such as BCL2, BCL-xL and MCL1 allows cancer cells to evade programmed cell death, leading to uncontrolled proliferation. Conversely, mutations or deletions in pro-apoptotic members such as BAX and BAK1 can further impair apoptosis. Understanding these molecular imbalances is essential to understanding how tumors develop resistance to chemotherapy and targeted therapies.
Cell Death Studies
In addition to cancer, apoptosis plays a fundamental role in many diseases, including neurodegenerative, autoimmune and cardiovascular diseases. Studying the molecular pathways regulated by the BCL2 family provides insight into how cell death contributes to disease progression. For example, excessive apoptosis is implicated in Alzheimer's and Parkinson's diseases, while impaired apoptosis underlies conditions such as autoimmune diseases. Research in this area is helping to identify potential therapeutic interventions that either enhance or suppress apoptosis to restore cellular balance in disease states.
Biomarker Development
The expression levels and mutations of BCL2 family members serve as valuable biomarkers for cancer prognosis and treatment response. For example, high BCL2 expression in chronic lymphocytic leukemia (CLL) is associated with poor prognosis. Similarly, BAX and BAK1 mutations can predict chemotherapy resistance in several cancers. Identification and validation of BCL2-related biomarkers will facilitate precision medicine by enabling patient stratification, guiding therapeutic decisions, and more effectively monitoring disease progression.
Drug Discovery
Given the critical role of BCL2 family proteins in apoptosis evasion, they have become prime targets for anticancer drug development. Small molecule inhibitors have shown significant success in the treatment of hematologic malignancies by selectively inhibiting BCL2, thereby restoring apoptotic sensitivity in cancer cells. Drug discovery efforts continue to focus on the development of new BCL2 inhibitors with improved efficacy and reduced resistance. High-throughput screening of novel compounds, structure-based drug design and combination therapy strategies are actively pursued to improve treatment outcomes in solid and hematological cancers.
Product Features
-
High Purity and Activity: Our recombinant proteins undergo rigorous quality control, ensuring exceptional purity and biological activity for reliable experimental outcomes.
-
Wide Selection: We offer an extensive range of recombinant proteins and antibodies, encompassing all major BCL2 family members for comprehensive research.
-
Custom Services: Creative BioMart offers tailored protein production and assay development services, designed to meet your specific research requirements and objectives.
-
Batch Consistency: Our stringent manufacturing processes ensure reproducible results across experiments by maintaining consistent quality in every production batch.
-
Advanced CRO Services: We offer comprehensive contract research services, including protein engineering and assay development, to accelerate your scientific discoveries.
Case Study
Case 1: BCL-XL Regulates Mitotic Apoptosis
Yu et al., 2024. BCL-XL regulates the timing of mitotic apoptosis independently of BCL2 and MCL1 compensation.
Mitotic catastrophe, induced by prolonged mitotic arrest, is an important anticancer strategy. BCL-xL, a key anti-apoptotic protein, regulates apoptosis during mitotic arrest, but its expression often adapts in response to downregulation or overexpression, complicating studies. This research found that BCL-xL and BCL2 work together to prevent apoptosis during mitotic arrest in epithelial cells such as HeLa and RPE1. Although BCL2 is more abundant, BCL-xL plays a more prominent role in suppressing apoptosis, particularly during mitotic arrest. Depletion of BCL-xL led to mitotic cell death by BAX and stabilized MCL1, which helped buffer apoptosis. These findings highlight the critical role of BCL-XL in regulating mitotic apoptosis.
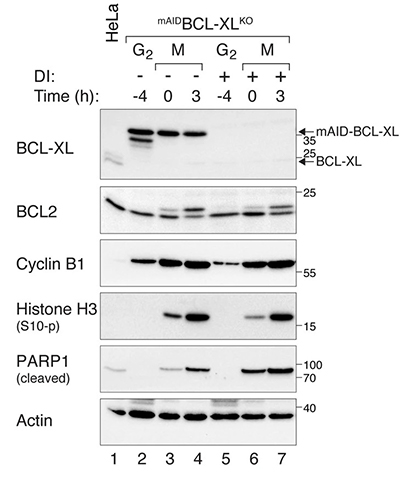
Figure 1. Increased PARP1 cleavage during mitotic arrest in BCL-XL-deficient cells. mAIDBCL-XLKO cells were synchronized and arrested in mitosis. Protein expression was analyzed with immunoblotting.
Case 2: Subcellular Localization of BCL2
Lalier et al., 2021. TOM20-mediated transfer of BCL2from ER to MAM and mitochondria upon induction of apoptosis.
This study investigates the subcellular localization of BCL2, a key antiapoptotic protein. In U251 glioma cells, BCL2 is primarily located in the ER, but translocates to mitochondria and MAM upon apoptosis induction. Mitochondrial translocation is regulated by TOM20, a protein involved in mitochondrial import. Interaction with TOM20 enhances the proapoptotic role of BCL2. Experiments with targeted BCL2 constructs show that ER-localized BCL2 protects against apoptosis, whereas mitochondria-targeted BCL2 fails to do so and even induces apoptosis. These findings highlight the complex role of BCL2 localization in apoptosis regulation and emphasize its interaction with TOM20 as a key apoptotic control mechanism.
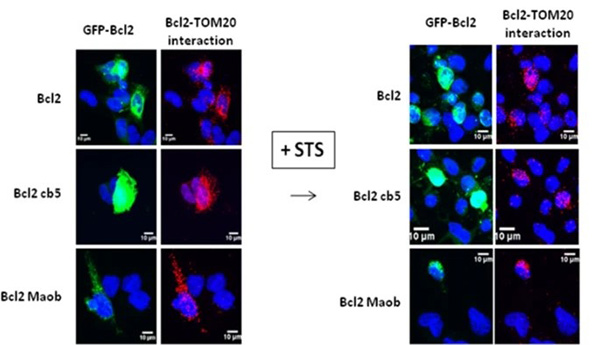
Figure 2. Transfected U251 cells were treated by STS and Bcl2–TOM20 interaction was measured by PLA.
FAQs
-
Q: What are BCL2 family proteins, and why are they important in research?
A: BCL2 family proteins are key regulators of apoptosis, the process of programmed cell death that is critical for maintaining cellular balance. They include both anti-apoptotic proteins such as BCL2 and BCL-xL, which promote cell survival, and pro-apoptotic proteins such as BAX and BAK, which facilitate apoptosis. Studying these proteins is critical to understanding the mechanisms underlying cancer, neurodegenerative diseases and immune disorders. -
Q: What types of BCL2 family products do you offer?
A: We provide a comprehensive range of products related to the BCL2 family, including:
- Recombinant Proteins: High-purity, biologically active proteins for functional and structural studies.
- Antibodies: Monoclonal and polyclonal antibodies suitable for various applications such as Western blotting and immunohistochemistry.
- Cell & Tissue Lysates: Human BCL2 family protein lysates derived from overexpression systems, prepared in RIPA buffer and supplied in SDS loading buffer—ready-to-use for downstream assays.
- Custom Services: Tailored protein production, assay development, and screening services to meet specific research needs.
-
Q: Can you help with custom projects involving BCL2 family proteins?
A: Absolutely. Our custom services include custom protein production, assay development and screening services. Our team of experts will work closely with you to develop solutions that meet your specific research goals.
-
Q: How do you ensure the quality of your BCL2 family products?
A: Our products undergo stringent quality control measures to ensure high purity and biological activity. We use advanced manufacturing processes and rigorous testing protocols to maintain batch-to-batch consistency, providing reliable and reproducible results for your experiments.