
Immunohistochemistry protocol
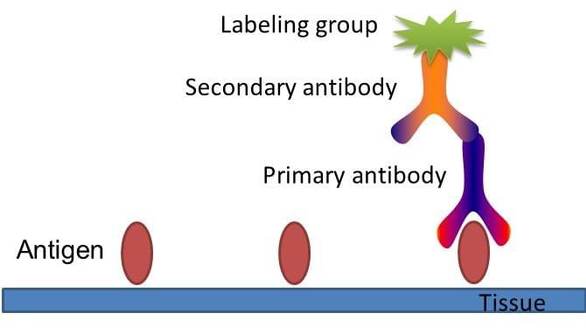
Fig. The schematic diagram of immunohistochemistry.
Here we provide a protocol for usual immunohistochemistry.
Fix
- Tissue samples are dissected from animals follow an appropriate protocol.
- Samples are immediately fixed in Carnoy’s solution and left overnight.
Embed
- The following day, the tissues are embedded into paraffin. To do so, each sample is washed with ethanol 4X for 45 minutes each time to remove the chloroform.
- Each sample is cleaned with xylene two times for 30 minutes each time at 58°C.
- The samples are infiltrated with paraffin 3 times for 20 minutes each at 58°C.
- The samples are then stored at room temperature overnight so that they would be ready the next day for sectioning.
Section
For immunofluorescence labeling of organ tissue, a paraformaldehyde fixation and paraffin embedding protocol is used. The organs are obtains by dissection.
- After the tissues are embedded in paraffin, a few blocks are placed in the freezer so they will be easier to section, and to avoid breaking the blocks.
- After 15 minutes in a freezer, the blocks are sectioned into 5 µm sections with the microtome and mounted onto slides.
- These are left overnight to dry.
Rehydration
- Slides are placed into the oven for 30 minutes.
- Slides are placed in xylene for 2 minutes (X2) in order to dissolve the paraffin.
- The rehydration process is then begun using 100% ethanol for 1 minute, then 95% ethanol for 1 minute, than 70% ethanol for 1 minute, and finally 50% ethanol for 1 minute.
Enzyme reactions
- Then, slides are washed with PBS/Tween 20 for 5 minutes and the slides are then dried and marked with the pap pen in order to keep the future solutions on the slide.
- 200 µL of PBS/proteinase kinase (PBS-PK) solution is applied onto each slide.
- The slides are incubated in a closed box for 15 minutes, and are then washed with PBS/Tween 20 for 5 minutes.
- Slides are then placed in peroxidase solution for 10 minutes (90% methanol/20% hydrogen peroxide) to open up the cell membranes.
Primary antibody binding
- The slides are once again washed with PBS/Tween 20 for 5 minutes (3 times).
- They are then placed inside a box to which water and paper are added to maintain moisture.
- A Zymed IHC kit is used and reactions are performed following manufacturer’s specifications as follows. A serum blocking solution is applied for 10 minutes. The primary antibody is prepared at dilutions of 1:100 and 1:2,000 respectively, and 200 µL of each primary antibody applies to the respective slides.
- These are incubated at 4°C freezer overnight.
Secondary antibody binding and coloration
- The next day, the slides are rinsed using PBS/Tween 20 for 2 minutes (3 X).
- Two drops of secondary antibody is then applied to each slide.
- After incubating for 10 minutes, the slides are rinsed with PBS/Tween 20 2 minutes (3 X).
- The AEC chromogen is then used follow the manufacturer’s protocols.
- After incubation, the slides are rinsed with distilled water, mounted, and pictures are taken.