



Computer-aided Enzyme Design
Creative BioMart offers comprehensive Computer-aided Enzyme Design services to accelerate protein and enzyme discovery through advanced computational approaches. Our team of highly skilled scientists, equipped with extensive academic and industrial experience, leverages state-of-the-art modeling software and powerful computing systems to deliver reliable and innovative design solutions. From ligand-based and structure-based approaches to pharmacophore elucidation and virtual screening, our services support both integrated medicinal chemistry projects and stand-alone programs. By combining expertise, technology, and collaboration, we help researchers shorten development timelines and optimize enzymes for diverse applications in pharmaceuticals, biotechnology, and industrial processes.

Introduction to Computer-Aided Enzyme Design and Engineering
The design and optimization of enzymes are critical in modern biotechnology and pharmaceutical research. Traditional experimental methods are often time-consuming and resource-intensive, limiting the pace of discovery. Computer-aided enzyme design (CAED) has emerged as a transformative approach, enabling researchers to explore enzyme structures, predict protein-ligand interactions, and evaluate functional properties with high precision.
By integrating computational modeling, virtual screening, and chemoinformatics, CAED provides an efficient pathway to identify new enzymes, improve catalytic efficiency, tailor substrate specificity, and optimize stability. When combined with experimental validation, this approach dramatically accelerates the design cycle, reduces costs, and enhances the likelihood of successful outcomes.

Figure 1. Computational enzyme engineering pipelines. Module 1: structure–function analysis to identify active site and substrate-binding pocket. Module 2: building enzyme–substrate complexes with molecular docking approaches. Module 3: identification of design positions for the subsequent sequence design. Module 4: engineering stability of enzymes. Module 5: engineering activity and specificity of enzymes. Module 6: screening for stability, affinity, and activity changes. (Scherer et al., 2021)
Computational Enzyme Design Services & Capabilities
Computational Enzyme Design Services
Creative BioMart provides end-to-end computer-aided enzyme design solutions, leveraging state-of-the-art modeling tools and structural biology expertise. Our services are tailored to accelerate enzyme engineering, optimize catalytic efficiency, and support research programs focused on biocatalysis and therapeutic enzyme development. Core services in this category include:
-
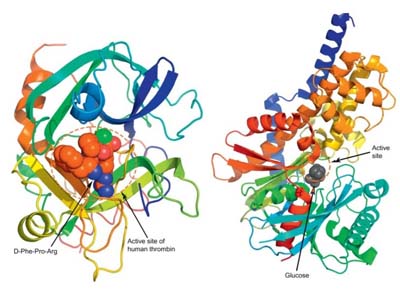
Conformational Analysis
-
Docking Studies
-
Homology Modeling
-
Structure-Based Design
Expanded Computational Drug Design Services
In addition to enzyme-focused design, our experienced scientists also provide computational drug discovery support, integrating ligand- and structure-based approaches to complement medicinal chemistry programs. These services help streamline hit identification, optimize lead compounds, and predict pharmacological properties. Our extended offerings include:
-
Compound Library Design and Database Management
-
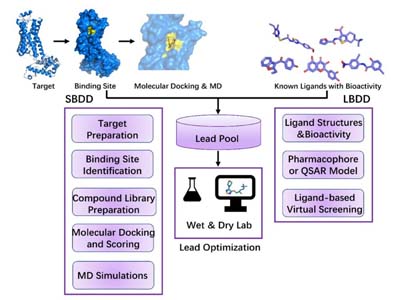
Structure-Based and Ligand-Based Drug Design
-
Virtual Screening for Hit Generation
-
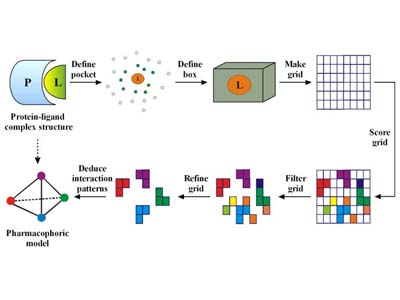
Pharmacophore Elucidation
-
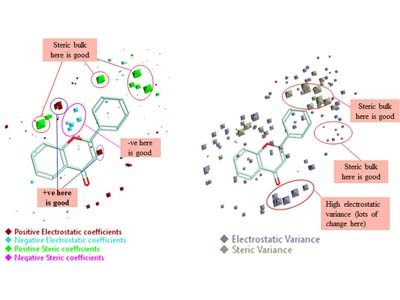
QSAR Analysis of Binding Affinity
-
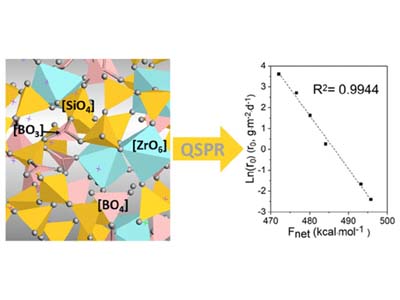
QSPR Analysis of ADME/Tox Properties
Our Capabilities
Our computer-aided enzyme design capabilities include:
| Capabilities |
Details |
|---|---|
|
Expert Scientists |
A multidisciplinary team of modelers with top research and industrial experience. |
|
Cutting-Edge Tools |
Access to state-of-the-art software and hardware, including:
|
|
Broad Application Range |
From drug discovery and enzyme engineering to biocatalysis and industrial biotechnology. |
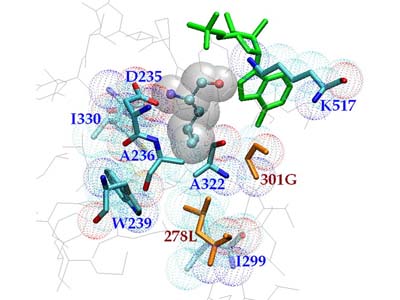
These enzymes are biologically and therapeutically relevant, with misregulation linked to cancer, neurodegeneration, and metabolic diseases. Our platform allows identification of potent inhibitors, including isoform-selective compounds critical for precision therapies.
Service Workflow

Why Partner with Creative BioMart
- Proven Expertise: A team of highly experienced modelers with both academic and industrial backgrounds.
- Advanced Technology: Equipped with industry-standard software and powerful computational infrastructure.
- Comprehensive Solutions: Ligand-based and structure-based approaches tailored to client needs.
- Cross-disciplinary Collaboration: Seamless integration with medicinal chemistry and experimental validation teams.
- Cost- and Time-efficient: Streamlined workflows reduce development timelines and resource expenditure.
- Global Perspective: A diverse team combining domestic and international experience ensures innovative problem-solving.
Real-World Applications of Computational Enzyme Engineering
Case 1: Nano-QSAR modeling of enzyme inactivation by metal oxide nanoparticles
Sizochenko et al., 2017. doi:10.3390/nano7100330
A quantitative structure–activity relationship (nano-QSAR) model was developed to explore how 24 metal oxide nanoparticles affect the activity of zebrafish ZHE1 enzyme. Using four key descriptors—hydrodynamic radius, mass density, Wigner–Seitz radius, and covalent index—the model, built with the M5P regression tree algorithm, demonstrated strong robustness (R2bagging = 0.90) and high external predictivity (Q2EXT = 0.93). Results aligned with current aquatic toxicity theories, showing dissolution and size-dependent properties as major drivers of enzyme inactivation. ZnO, CuO, Cr2O3, and NiO nanoparticles exhibited strong inhibitory effects, and the model provides a non-experimental alternative to animal testing.
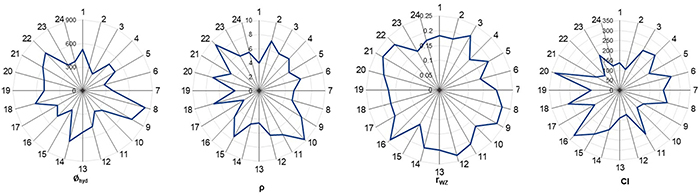
Figure 2. Radar plot of the distribution of used descriptors. (Sizochenko et al., 2017)
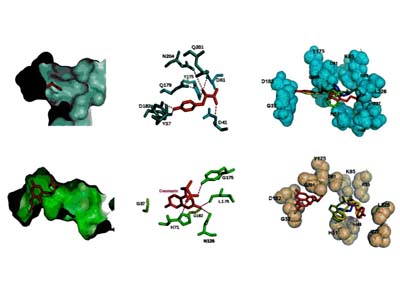
Case 2: Virtual screening against metalloenzymes for inhibitors and substrates
Irwin et al., 2017. doi:10.1021/bi050801k
Molecular docking leverages the three-dimensional structure of a receptor to screen small-molecule databases for potential ligands, but metalloenzymes pose challenges due to partial covalent metal–ligand interactions. Using a standard scoring function, researchers docked 95,000 compounds from the MDL Drug Data Report against five metalloenzymes, achieving at least 20-fold enrichment of annotated ligands. Prospective screening of a zinc β-lactamase identified five inhibitors ( Ki < 120 μM), including a 2 μM lead, while substrate prediction for a Zn-dependent phosphotriesterase successfully confirmed eight previously uncharacterized substrates. These results demonstrate that noncovalent docking can effectively identify inhibitors and substrates of certain metalloenzymes.
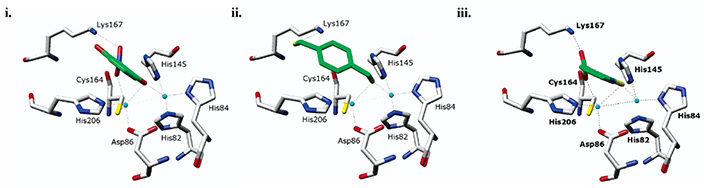
Figure 3. Docked geometries of top scoring hits against the metallo-β-lactamase from Bacteroides fragilis. Metal atoms are cyan spheres. Distances from ligand to zinc atoms and Lys NZ shown. (Irwin et al., 2017)
Client Feedback on Our Enzyme Design and Drug Discovery Support
"We collaborated with Creative BioMart to identify inhibitors for a key metabolic enzyme in our oncology program. Their computer-aided enzyme design team used ligand-based virtual screening and pharmacophore elucidation to narrow thousands of compounds down to a focused hit list. Several of these hits translated into confirmed actives during our in-house assays, saving us months of early discovery work. Their reporting was detailed, reproducible, and easy to integrate into our pipeline."
— Director of Discovery Chemistry | Mid-Sized Pharmaceutical Company
"Our company required an engineered enzyme with improved thermal stability for large-scale biocatalysis. Creative BioMart applied structure-based modeling, conformational analysis, and homology modeling to predict mutations enhancing stability without sacrificing activity. The optimized variants they proposed showed a 3-fold increase in half-life at elevated temperatures, leading to significant cost savings in production. Their combination of computational rigor and practical insight exceeded our expectations."
— Head of Bioprocess Development | Global Industrial Biotech Company
"Creative BioMart played an essential role in our drug development program targeting a neurological enzyme. Their scientists combined QSAR analysis of binding affinity with QSPR predictions of ADME/Tox properties, allowing us to prioritize candidates with both strong activity and favorable drug-like profiles. The ability to balance potency with pharmacokinetics early on de-risked our project and streamlined our preclinical efforts. Their expertise was evident in every stage of the collaboration."
— Senior Program Manager, CNS Research | International Pharmaceutical Company
"As an academic lab working on novel enzyme functions, we lacked the computational resources to explore enzyme-ligand interactions at scale. Creative BioMart provided docking studies, homology models, and compound database management tailored to our project. Their team helped us rationalize our mutagenesis strategy, which led to the publication of a high-impact paper on enzyme specificity. Their willingness to adapt to our unique research needs made them an outstanding partner."
— Principal Investigator, Biochemistry Department | Leading Research University
FAQs About Computational Enzyme Design Services
-
Q: What is computer-aided enzyme design, and why should I use it?
A: Computer-aided enzyme design (CAED) uses advanced computational modeling and simulation to predict enzyme structure, activity, and stability. It accelerates discovery by reducing the need for trial-and-error experiments, saving both time and resources. This approach is ideal for drug discovery, biocatalysis, and enzyme engineering projects. -
Q: What kinds of projects can you support with CAED?
A: We support a wide range of projects, including:- Hit identification and lead optimization in drug discovery
- Enzyme stability and activity optimization for industrial applications
- Structure-based ligand design and docking studies
- Pharmacophore elucidation and conformational analysis
- Early-stage ADME/Tox profiling using QSPR models
-
Q: What tools and technologies do you use for enzyme design?
A: Our team uses state-of-the-art software and hardware platforms, including the Schrödinger package, Accelrys package, 3D stereo graphic systems, chemoinformatics tools, and database packages. These enable us to deliver highly accurate modeling, virtual screening, and predictive analyses. -
Q: How experienced is your team?
A: Our scientists have extensive experience in both academic and industrial environments. The team includes internationally trained modellers, ensuring a global perspective combined with practical, real-world expertise. -
Q: Can you help with inhibitor screening and selectivity assessment?
A: Yes. Our ligand-based and structure-based approaches, combined with pharmacophore and docking studies, are well suited for high-throughput virtual screening and selectivity evaluation. This capability reduces the risk of late-stage drug failure and supports more cost-efficient development.
Resources
Related Services
- Protein Interaction Service
- Ligand-Protein Interactions Screening Based on Gold Nanoparticles
- Protein-Lipids/Nucleic Acid Interaction Assay and Screening
- Protein Sequence Analysis and Function Prediction
- Drug Discovery Screening
- Protein Engineering Services
- Sequence Based Protein Design
- Structure Based Protein Design
Related Products
References:
- Alam S, Khan F. 3D-QSAR, Docking, ADME/Tox studies on Flavone analogs reveal anticancer activity through Tankyrase inhibition. Sci Rep. 2019;9(1):5414. doi:10.1038/s41598-019-41984-7
- Bhagavan NV, Ha CE. Enzymes and enzyme regulation. In: Essentials of Medical Biochemistry. Elsevier; 2015:63-84. doi:10.1016/B978-0-12-416687-5.00006-3
- Chen CY, Georgiev I, Anderson AC, Donald BR. Computational structure-based redesign of enzyme activity. Proc Natl Acad Sci USA. 2009;106(10):3764-3769. doi:10.1073/pnas.0900266106
- Cheng T, Liu Z, Wang R. A knowledge-guided strategy for improving the accuracy of scoring functions in binding affinity prediction. BMC Bioinformatics. 2010;11(1):193. doi:10.1186/1471-2105-11-193
- Irwin JJ, Raushel FM, Shoichet BK. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemi stry. 2005;44(37):12316-12328. doi:10.1021/bi050801k
- Lu X, Deng L, Gin S, Du J. Quantitative structure–property relationship (QSPR) analysis of zro2 -containing soda-lime borosilicate glasses. J Phys Chem B. 2019;123(6):1412-1422. doi:10.1021/acs.jpcb.8b11108
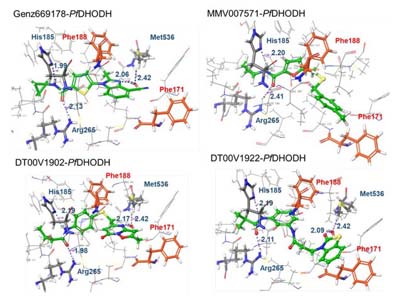
- Rawat R, Verma SM. High-throughput virtual screening approach involving pharmacophore mapping, ADME filtering, molecular docking and MM-GBSA to identify new dual target inhibitors of Pf DHODH and Pf Cytbc1 complex to combat drug resistant malaria. Journal of Biomolecular Structure and Dynamics. 2021;39(14):5148-5159. doi:10.1080/07391102.2020.1784288
- Scherer M, Fleishman SJ, Jones PR, Dandekar T, Bencurova E. Computational enzyme engineering pipelines for optimized production of renewable chemicals. Front Bioeng Biotechnol. 2021;9:673005. doi:10.3389/fbioe.2021.673005
- Sizochenko N, Leszczynska D, Leszczynski J. Modeling of interactions between the zebrafish hatching enzyme ZHE1 and a series of metal oxide nanoparticles: nano-QSAR and causal analysis of inactivation mechanisms. Nanomaterials. 2017;7(10):330. doi:10.3390/nano7100330
- Steinberg X, Galpin J, Nasir G, et al. A rationally designed aminoacyl-tRNA synthetase for genetically encoded fluorescent amino acids. Preprint posted online July 22, 2016. doi:10.1101/065227
- Wassermann AM, Camargo LM, Auld DS. Composition and applications of focus libraries to phenotypic assays. Front Pharmacol. 2014;5. doi:10.3389/fphar.2014.00164
- Zhang Y, Luo M, Wu P, Wu S, Lee TY, Bai C. Application of computational biology and artificial intelligence in drug design. IJMS. 2022;23(21):13568. doi:10.3390/ijms232113568
Contact us or send an email at for project quotations and more detailed information.
Quick Links
-

Papers’ PMID to Obtain Coupon
Submit Now -

Refer Friends & New Lab Start-up Promotions