
Super Gene Family
-
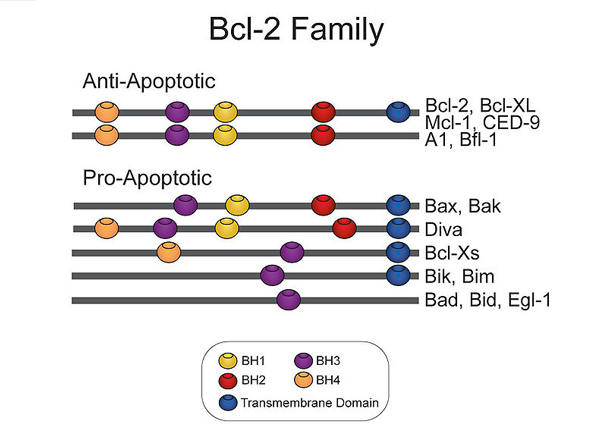
Bcl2 Gene Family
Proteins of the BCL2 family govern whether a cell lives or commits to apoptosis through the intrinsic apoptotic pathway...
-
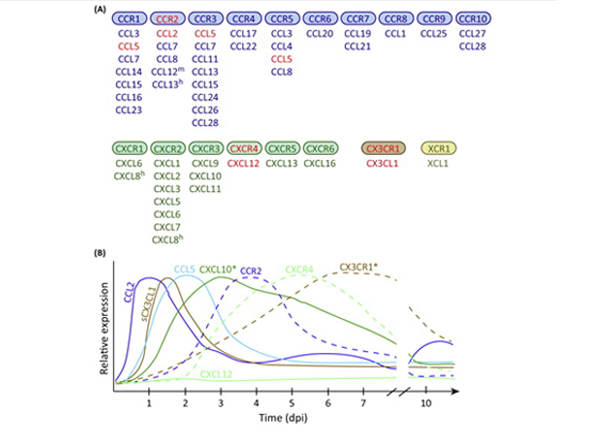
Chemokine
The chemokine family of proteins has broad, diverse functional repertoires. However, their structural variation is narrow...
-
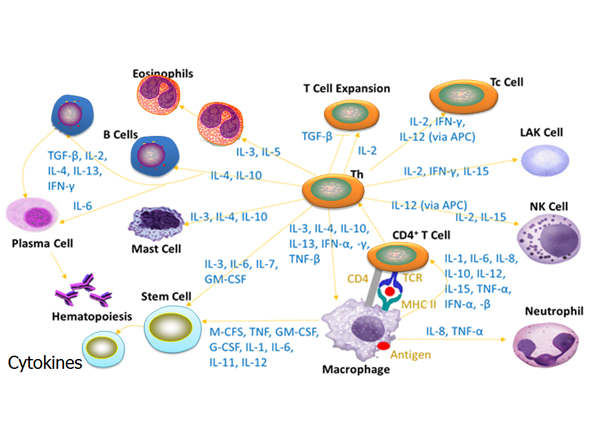
Cytokines
Cytokines are small, water-soluble, signaling proteins and glycoproteins secreted by hematopoietic and non-hematopoietic...
-
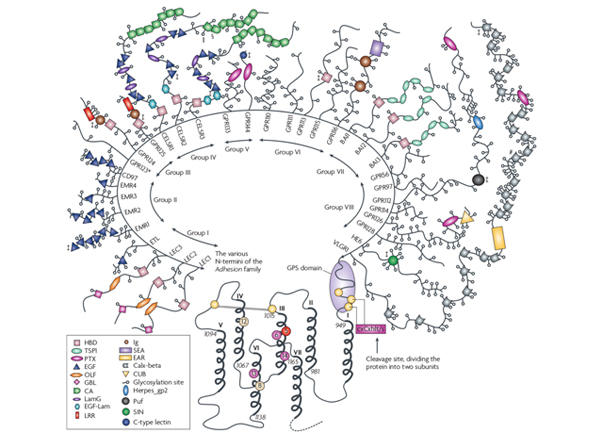
GPCRs
G-protein coupled receptors (GPCRs) are cell surface receptors that play a critical role in cell signaling...
-
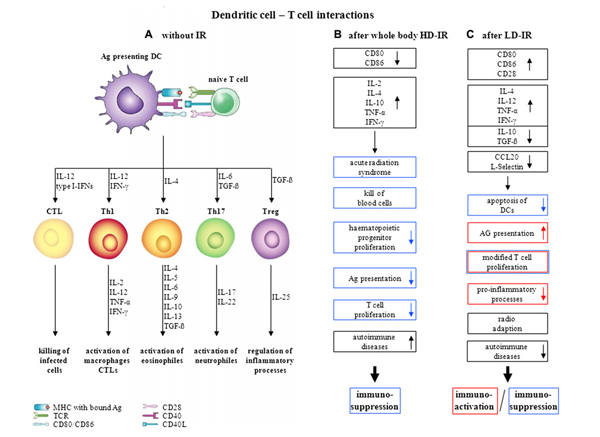
IL
IL superfamily, a member of larger group cytokines, is consisting of proteins called interleukins and interleukin receptors...
-
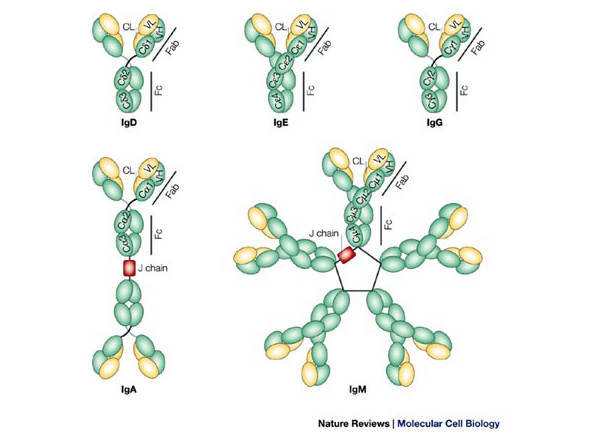
Immunogloblulin
The immunoglobulin superfamily (IgSF) is a class of proteins that are associated with the adhesion, binding and recognition processes of cells...
-
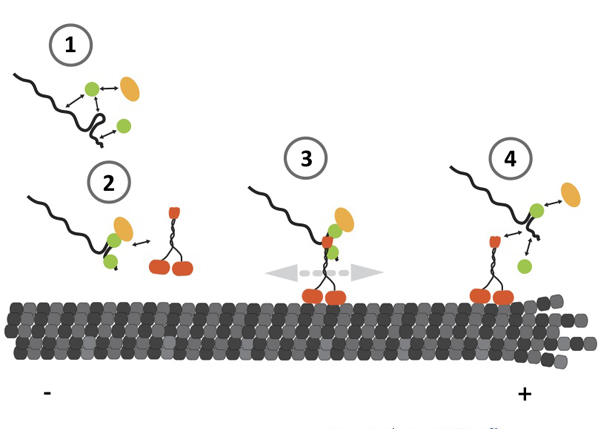
Kinesins(KIF)
Genes in the Kinesins Gene Family (KIF) superfamily code proteins called kinesins. There are 46 different kinesins in humans and other mammals...
-
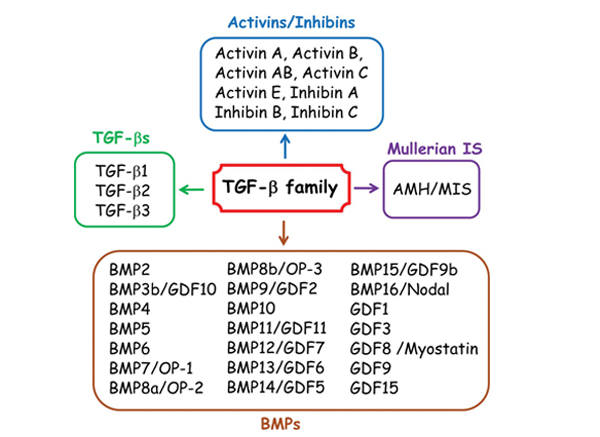
TGF
The transforming growth facor beta (TGF-β) superfamily is a large family of growth factors named after the first member TGF-β1...
-
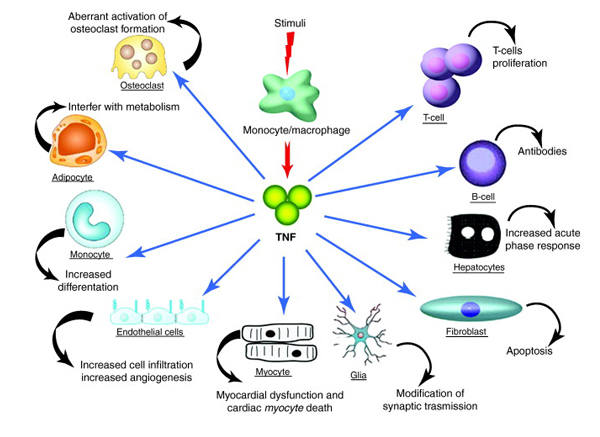
TNF
Tumor necrosis factor (TNF) superfamily composes of a group of cytokines and their receptors which can reduce cell death (apoptosis)...
-
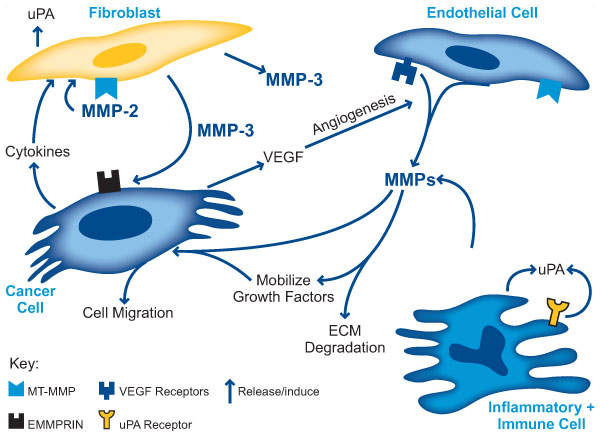
MMPs
The matrix metalloproteinases (MMPs) are one of the major families of proteinases that play key roles in the responses of cells to...
-
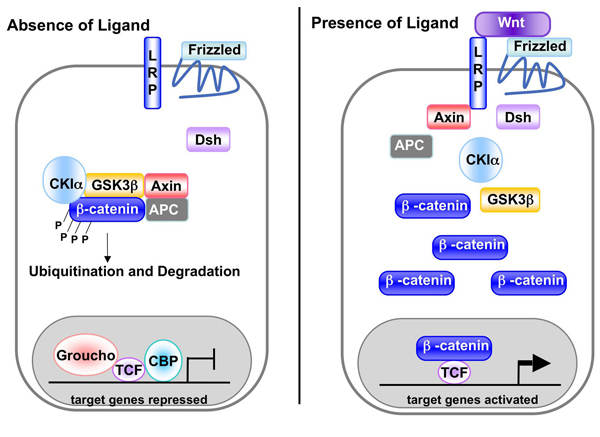
WNT
Wnt proteins are characterized by the presence of a hydrophobic signal sequence followed by an invariant spacing pattern of 22 highly conserved...
-
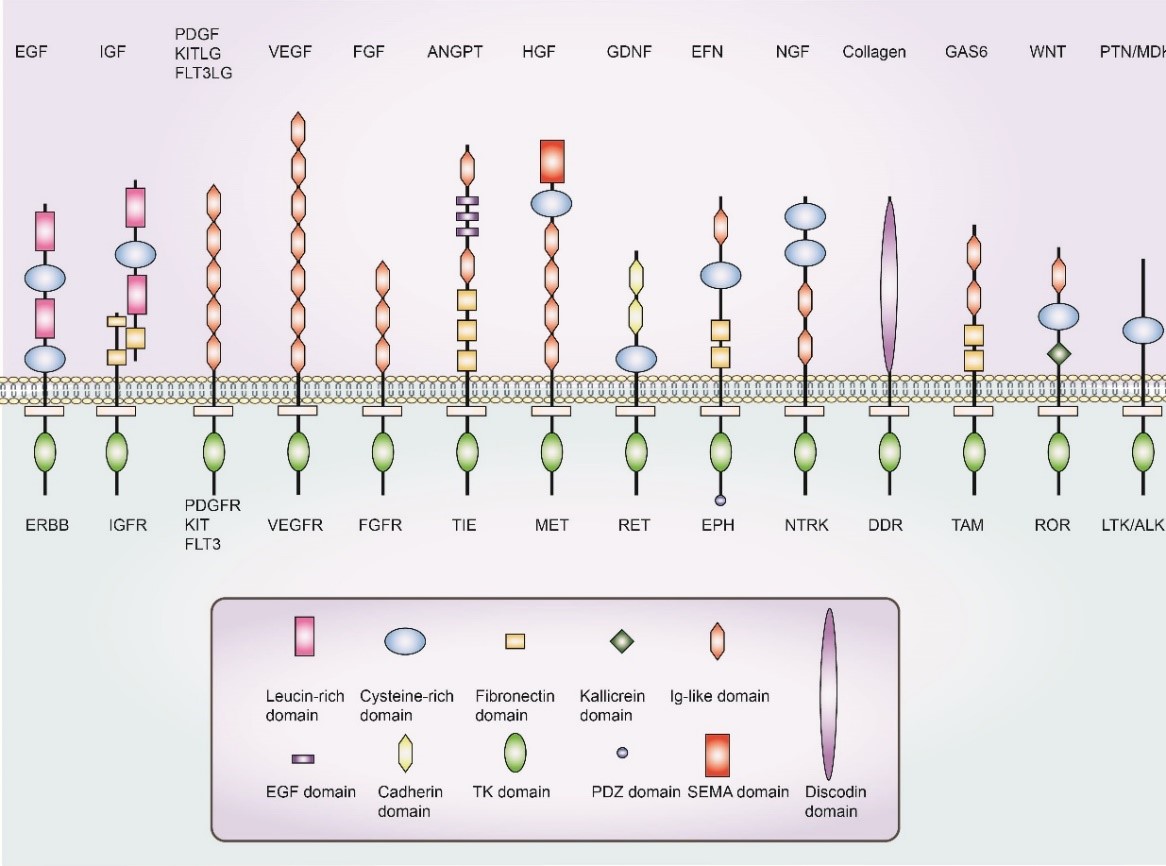
Growth Factors
Growth factors are a class of organic substances that are essential for regulating the normal growth and metabolism of microorganisms but cannot....
Contact us or send an email at for project quotations and more detailed information.
Quick Links
-

Papers’ PMID to Obtain Coupon
Submit Now -

Refer Friends & New Lab Start-up Promotions