
A Reliable Recombinant Proteins Supplier
The application of recombinant proteins:
To identify the polypeptide coded by a DNA-sequence
To analyze the biological activity
To study the structure-function relationships, interactions
To study the 3D-structure
To do protein engineering and design
To raise specific antibodies
To develop a target-specific drug
To produce therapeutic proteins
To produce vaccines
To produce biotechnological enzymes
How to make:
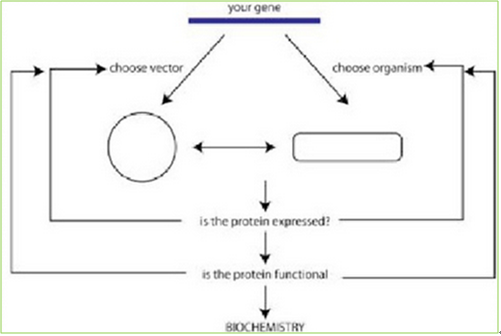
The factors that determine the selection of protein production host:
Produced protein-prokaryotic/eukaryotic; cytoplasmic/secreted; organelle specific;
Amounts needed-analytical, functional studies (ng); antibody production (μg-mg); structural biology (mg); coomercial (g-kg);
Demands on authenticity-active/inactive=native/denatured; modified/not modified (glycosylation etc.); molecular weight; sequence; increased expression= decreased authenticity;
Money;
Protein production;
Difference for various hosts:
For human proteins, mammalian cells may be the best, but they are also the slowest and most expensive to use.
Insect cells (baculovirus) have many of the advantages of mammalian cell culture (post-translational modifications, eukaryotic cell system, etc.) but they are faster and cheaper to use.
Yeast is an option for some things, but the differences can bite (particularly in glycosylation pattern and extraction).
E. coli is the cheapest, easiest and fastest by far, but it just doesn’t work for some proteins.
Do you need to add co-factors to the media? Do you need a chaperone or second protein for correct folding?
With over 20,000 recombinant proteins, Creative BioMart provides highly expressed recombinant proteins in various hosts to meet customers’ requirements, such as CHO Cells, E. coli, Insect Cells, Mammalian Cells, Nicotiana benthamiana, Virus, Yeast and other expression systems. For specific products, visit http://www.creativebiomart.net/producttypelist_1.htm or call us today.
Article Link: A Reliable Recombinant Proteins Supplier