
Protein Purification Instruction
By analyzing pure protein, we can determine amino acid sequences and investigate a protein’s biochemical function. Besides, crystallization of proteins need pure protein, and we can obtain x-ray data from the protein crystals with a picture of tertiary structure, which is the actual functional unit of the protein. For pure protein is so important for further analysis, we will discuss the protein purification methods which usually used.
First, there is a problem, how could we know the purified protein is what we are looking for?
Purified protein contains only one type of protein which we are interested in. At beginning, this protein may be only a tiny fraction of the complex, how could we know we isolate the particular protein we need?
We need an assay for some unique identifying property of the protein, so that we can tell when the protein is present or whether this is the target protein. Determining an effective assay is often difficult; but the more specific the assay, the more effective the purification. For enzymes, which are protein catalysts, the assay is usually based on the reaction that the enzyme catalyzes in the cell. To be certain that our purification scheme is working, we need one additional piece of information—the amount of protein present in the mixture being assayed. There are various rapid and accurate means of determining protein concentration. For already known protein, there is a more convenient method-western blot. We can use specific antibody to analyze the target protein. If the target protein does not has suitable antibody, for recombinant protein, the antibody of co-expressional tag could also be used for analysis. But for native protein, you need to find other analysis method. These methods will be used after the purification process. We’ll talk about this later.
When you know how to analyze whether your purification process is successful. We can start to purify the protein now.
First, the protein need be released from cell. There are several methods to disrupt the cell membrane. Sonication (ultrasound producing cavitation) is a common method. Others include French press, enzymatic or chemical cleavage of the cells, and freeze–thawing. For larger scale continuous disruption, high pressure homogenization or bead milling are widely used.
After that step, the mixture is fractionated by centrifugation. Here, we need make some trials or dependent on previous experience to make sure the target protein enriched in which component of the fractions. The differential centrifugation yields a dense pellet of heavy material at the bottom of the centrifuge tube and a lighter supernatant above, then the supernatant is again centrifuged at a greater force to yield yet another pellet and supernatant. In the centrifugation process, the mixture yields several fractions of decreasing density, each still containing hundreds of different proteins, which are subsequently assayed for the further purification.
Proteins could be purified according to their basic characteristics, such as solubility, size, charge, and specific binding affinity. Usually, protein mixtures are subjected to a series of separations, each based on a different property to yield a pure protein.
Salting Out
Most proteins are less soluble at high salt concentrations, an effect called salting out. The salt concentration at which a protein precipitates differs from one protein to another. Hence, salting out can be used to fractionate proteins. Salting out is also useful for concentrating dilute solutions of proteins, including active fractions obtained from other purification steps.
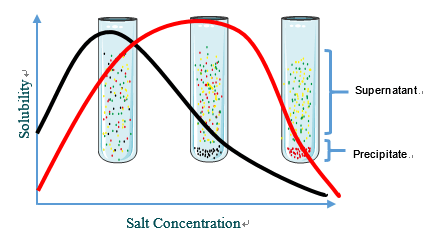 Fig1. Salting out
Fig1. Salting out
Dialysis
Dialysis can be used to remove the salt in above step. Proteins can be separated from small molecules by dialysis through a semipermeable membrane, such as a cellulose membrane with pores. Proteins have greater dimensions than the pore diameter and are retained inside the dialysis, whereas smaller molecules and ions traverse outside the bag through the pore. However, this process could not distinguish between proteins effectively.
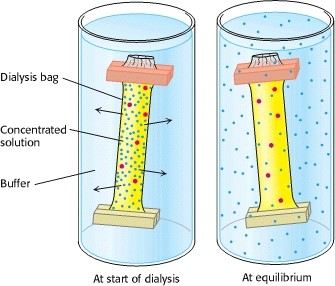 Fig2. Dialysis
Fig2. Dialysis
Chromatography is the more discriminating separations on the basis of size. The column consists of porous beads made of insoluble but highly hydrated polymer such as dextran or agarose or polyacrylamide. We applied sample on the top of a column, the small molecules can enter the porous of the beads, but large ones cannot. Thus, the small molecule goes both inside and between the beads, whereas large one goes only between the beads. Small molecules flow longer distance than the larger molecules. So, the large molecules flow more rapidly through this column and emerge first. Molecules that are of a size to occasionally enter a bead will flow from the column at an intermediate position, and small molecules, which take a longer, tortuous path, will exit last.
%20Gel-Filtration%20Chromatography%20and%20(B)%20Ion-Exchange%20Chromatography.png) Fig3. (A) Gel-Filtration Chromatography and (B) Ion-Exchange Chromatography
Fig3. (A) Gel-Filtration Chromatography and (B) Ion-Exchange Chromatography
Proteins may have net positive or negative charge at different pH range. This is another basic rules for protein separate. If a protein has a net positive charge, it will usually bind to a column of beads containing carboxylate groups, whereas a negatively charged protein will bind to a column of beads containing diethylaminoethyl group. Protein sample loads on the column, a positively charged protein binds to the beads, and other charged protein will flow through this column and emerge first. The positively charged protein, for example, can ben eluted by increasing the concentration of sodium chloride or another salt in the eluting buffer because sodium ions compete with positively charged groups on the protein for binding to the column. Proteins with less net positive charge released first, then the higher charge density proteins. There are lots of ion-exchange chromatography, for example, CM cellulose resin, DEAE dextran resin, DEAE polystyrene resin, and so on. You need to choose suitable chromatography depends on the character of your interested protein.
Affinity Chromatography
Affinity chromatography is another powerful and generally applicable means of purifying proteins. This technique takes advantage of the high affinity of many proteins for specific chemical groups. For example, Ni polystyrene resin is widely used for the purification of his-tagged protein. His protein has affinity for Ni polystyrene. When passing a column containing Ni polystyrene, his-tagged protein attached to the modified resin, whereas most other proteins do not. For other native protein, for example, the plant protein concanavalin A can be purified by passing through a column of beads containing covalently attached glucose residues. There are also lots of affinity chromatography for target protein, but you should know the protein affinity character first, or you need some trials to find suitable one. Affinity chromatography is a powerful means of isolating transcription factors. A protein mixture is percolated through a column containing specific DNA sequences attached to a matrix; proteins with a high affinity for the sequence will bind and be retained. After that, the transcription factor could be washed by high concentration salt solution.
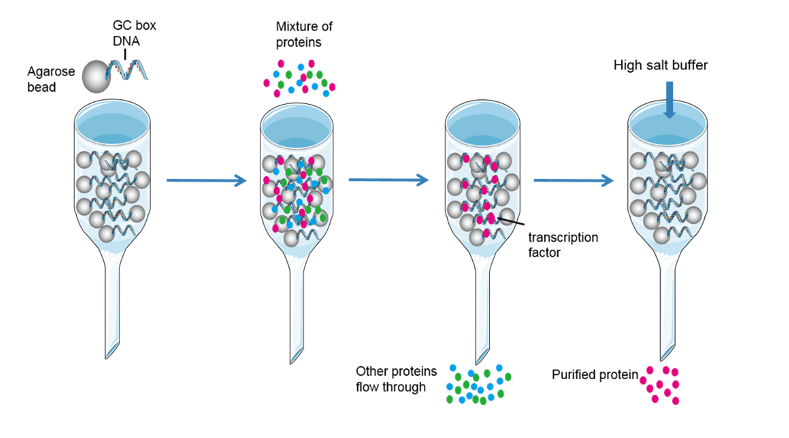 Fig4. Affinity chromatography
Fig4. Affinity chromatography
In conclusion, affinity chromatography isolate target protein in three steps. Firstly, attaching the chemical group which could affinity with target protein to the column. Secondly, loading the sample into the column, and let the target protein affinity with chemical group. Thirdly, eluting the target protein by high concentration salt solution or other method to decrease binding affinity.
High-Pressure Liquid Chromatography
The resolving power of all of the column techniques can be improved substantially through the use of a technique called high-pressure liquid chromatography (HPLC). This techniques could enhanced the ability of other columns discussed above. The column materials themselves are much more finely divided and, as a consequence, there are more interaction sites and thus greater resolving power. When using HPLC, the samples load into the column and move through the stationary phase under the pressure. For different material have different affinity with stationary phase, the objects leave the column in different time. The detector obtains various peak signal and each peak represents a different kind of compound. After comparing analysis of these signals, we will know the material contained in the sample. The net result is high resolution as well as rapid separation.
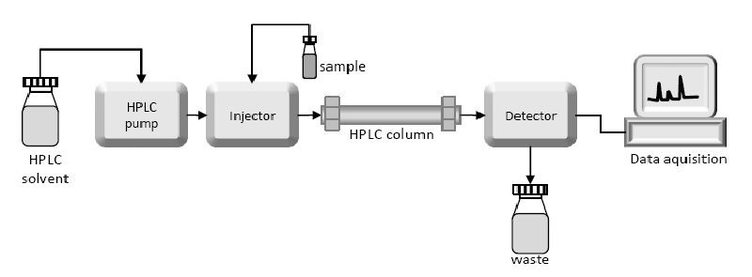 Fig5. High-Pressure Liquid Chromatography
Fig5. High-Pressure Liquid Chromatography
After all the purification steps, how could we know our purification scheme is effective? Apart from specific activity rises in each purification step, another method is using electrophoresis to display the target proteins at each step. Proteins can be separated largely on the basis of mass by electrophoresis in a polyacrylamide gel under denaturing conditions. SDS-polyacrylamide gel electrophoresis (SDS-PAGE) is rapid, sensitive, and capable of a high degree of resolution. We can examine the efficacy of our purification scheme by analyzing a part of each fraction by SDS-PAGE. The initial fractions will display dozens to hundreds of proteins. As the purification progresses, the number of bands will diminish, and the prominence of one of the bands should increase. This band will correspond to the protein of interest.
In case some protein may have similar weight with the target protein, we need to run isoelectric focusing first. Isoelectric focusing can be combined with SDS-PAGE to obtain very high resolution separations. Proteins can also be separated electrophoretically on the basis of their relative contents of acidic and basic residues. The isoelectric point (pl) of a protein is the pH at which its net charge is zero. Protein with similar weight are less possible have same pI, thus, using two-dimensional electrophoresis, proteins have been separated in the horizontal direction on the basis of isoelectric point and in the vertical direction on the basis of mass.
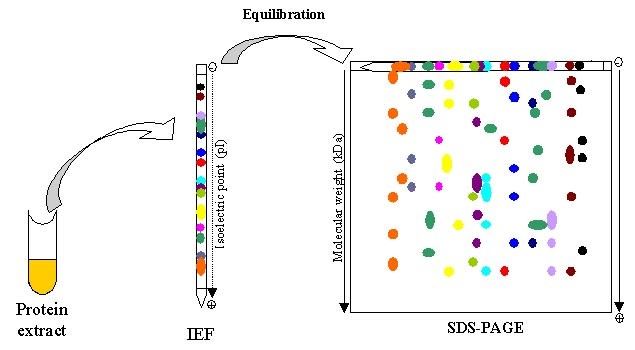 Fig6. Two-dimensional electrophoresis
Fig6. Two-dimensional electrophoresis
Besides quality analysis of protein purification, we also need quantify the target protein at each step. If the target protein is an enzyme, we could determine the protein concentration by enzyme activity. On the other hand, if the target protein doesn’t have the enzyme activity, we could semi-quantify the target protein through SDS-PAGE gel, which could analysis the percentage of the target protein in total sample. If we load a constant amount of protein onto each lane after each step, the number of bands decreases in proportion to the level of purification, and the amount of protein of interest increases as a proportion of the total protein present.
A good purification scheme takes into account both purification levels and yield. A high degree of purification and a poor yield leave little protein with which to experiment. A high yield with low purification leaves many contaminants (proteins other than the one of interest) in the fraction and complicates the interpretation of experiments.
Finally, mass spectrometry could determine the target protein mass more precisely. These methods are called matrix-assisted laser desorption-ionization (MALDI) and electrospray spectrometry. Using this method, we could learn more about our target protein.
We provide protein expression and purification services in our company. If you have any questions on protein purification, welcome to contact us for details!