
Coagulation
Creative BioMart Coagulation Product List
Immunology Background
About Coagulation
Coagulation, also known as blood clotting or hemostasis, is a complex physiological process that prevents excessive bleeding when blood vessels are damaged. It involves a series of sequential and tightly regulated events that lead to the formation of a blood clot, which helps to seal the injury site and initiate the repair process. The coagulation process can be broadly divided into three main stages: primary hemostasis, secondary hemostasis, and fibrinolysis.
- Primary Hemostasis: The initial phase of coagulation is primary hemostasis, which involves the formation of a platelet plug at the site of vascular injury. When a blood vessel is damaged, platelets are activated and adhere to the exposed collagen fibers in the vessel wall. This adhesion is facilitated by the interaction of platelet receptors, such as glycoprotein Ib (GPIb), with von Willebrand factor (vWF), a protein released by endothelial cells and platelets. Activated platelets then undergo shape change, release granules containing various molecules, and aggregate to form a platelet plug. This process is also supported by the activation of plasma clotting factors and the generation of thrombin.
- Secondary Hemostasis: Secondary hemostasis involves the coagulation cascade, a series of enzymatic reactions that amplify and stabilize the initial platelet plug. The coagulation cascade is initiated by the exposure of tissue factor (TF) at the site of injury, either from damaged cells or from the subendothelial matrix. TF forms a complex with factor VII, leading to the activation of factors IX and X. Activated factor X converts prothrombin to thrombin, which is a central enzyme in the coagulation process. Thrombin cleaves fibrinogen, a soluble plasma protein, into insoluble fibrin strands. The fibrin strands form a mesh-like network that traps platelets, red blood cells, and other components to form a stable blood clot.
- Fibrinolysis: Once the damaged blood vessel has been repaired, it is important to remove the blood clot and restore normal blood flow. Fibrinolysis is the process by which the clot is degraded. Plasminogen, a protein present in the blood, is converted to plasmin, an enzyme that breaks down fibrin. Plasmin is generated through the activation of plasminogen by tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA), which are released from endothelial cells and other sources. Plasmin cleaves fibrin into smaller fragments called fibrin degradation products, allowing the clot to dissolve.
Overall, coagulation is a dynamic and tightly regulated process that involves the interaction of platelets, plasma clotting factors, and the endothelium. It ensures that blood remains fluid under normal circumstances but rapidly forms a clot to prevent excessive bleeding in response to vascular injury. The balance between procoagulant and anticoagulant mechanisms is crucial to maintain hemostasis and avoid pathological conditions such as thrombosis or bleeding disorders.
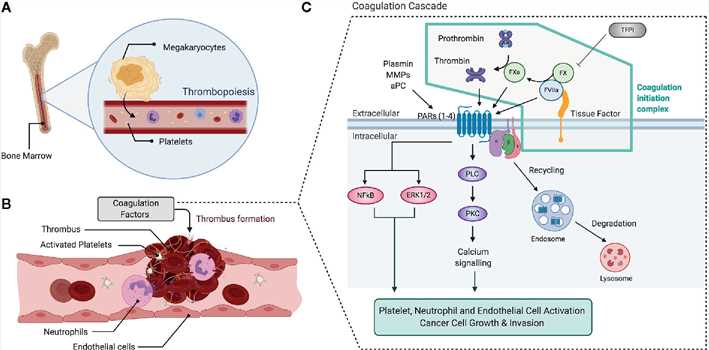 Fig.1 Overview of the coagulation and clotting factor signaling. (Hill CN, et al., 2020)
Fig.1 Overview of the coagulation and clotting factor signaling. (Hill CN, et al., 2020)
Molecules in the Coagulation Process
Coagulation Cascade Protease Inhibitors
- Antithrombin (AT): AT is a serine protease inhibitor that inhibits several coagulation factors, including thrombin (factor IIa), factor Xa, and other proteases involved in the clotting process.
- Protein C Inhibitor (PCI): PCI is a serine protease inhibitor that inhibits activated protein C, a natural anticoagulant that inactivates factors Va and VIIIa.
- Tissue Factor Pathway Inhibitor (TFPI): TFPI inhibits the tissue factor pathway by binding to factor Xa and the factor VIIa-tissue factor complex, thereby regulating the initiation of the coagulation cascade.
Coagulation Cascade Proteases
- CPB2 (Carboxypeptidase B2), also known as thrombin-activatable fibrinolysis inhibitor (TAFI), is produced in an inactive form (procarboxypeptidase B2) and requires thrombin or other proteases to cleave and activate it. Activated CPB2 functions as a carboxypeptidase, removing C-terminal lysine residues from partially degraded fibrin, thereby inhibiting plasminogen binding and slowing down the fibrinolysis process.
- KLKB1 (Kallikrein B1): KLKB1 is a protease that is involved in the activation of the kallikrein/kinin system. It cleaves kininogen to generate bradykinin, a potent vasoactive and proinflammatory peptide. KLKB1 is activated by factor XIIa, and once activated, it can further activate factor XI, leading to the amplification of the coagulation cascade.
- PLAT (Tissue Plasminogen Activator): PLAT, also known as tissue-type plasminogen activator (tPA), is a serine protease that plays a crucial role in fibrinolysis. It converts plasminogen, an inactive precursor, into plasmin, an active enzyme that breaks down fibrin clots. PLAT is primarily produced by endothelial cells and is released in response to various stimuli, such as vascular injury or thrombotic events.
Coagulation Factors
- Fibrinogen (Factor I): Fibrinogen is a soluble plasma protein that is converted to insoluble fibrin by the action of thrombin. Fibrin forms the structural framework of a blood clot.
- Factor V: Factor V is a cofactor that, together with factor Xa and calcium ions, forms the prothrombinase complex, which converts prothrombin to thrombin.
- Factor VIII: Factor VIII is a cofactor that, together with factor IXa, forms the tenase complex, which activates factor X in the coagulation cascade.
Other Coagulation Cascade Molecules
- von Willebrand Factor (vWF): vWF is a glycoprotein involved in primary hemostasis. It mediates platelet adhesion to damaged vascular walls and stabilizes factor VIII in the circulation.
- Phospholipids: Phospholipids, such as phosphatidylserine, are essential for the assembly and activation of coagulation factors on the platelet surface during clot formation.
- Calcium ions: Calcium ions play a crucial role in the coagulation cascade by facilitating the assembly and activation of coagulation factors.
- Platelet Membrane Receptors: Various receptors on platelets, such as glycoprotein Ib (GPIb) and integrins, mediate platelet adhesion, activation, and aggregation during clot formation.
Plasmin/Plasminogen and Kallikrein/Kinin Systems
- Plasmin: Plasmin is an enzyme that breaks down fibrin, leading to the dissolution of blood clots. It is derived from plasminogen through the action of tissue-type plasminogen activator (tPA) or urokinase-type plasminogen activator (uPA).
- Plasminogen: Plasminogen is a precursor protein that can be converted to plasmin. It circulates in the blood and is activated at the site of a clot by plasminogen activators, such as tPA and uPA.
- Kallikrein: Kallikrein is an enzyme involved in the activation of the kallikrein/kinin system. It cleaves kininogen to generate bradykinin, a potent vasoactive and proinflammatory peptide.
- Bradykinin: Bradykinin is a peptide generated by the action of kallikrein on kininogen. It causes vasodilation, increases vascular permeability, and promotes inflammation.
These molecules collectively contribute to the tightly regulated coagulation process, ensuring a balance between clot formation and clot dissolution, and maintaining hemostasis in the body.

Available Resources for Coagulation
- At Creative BioMart, we present an extensive array of coagulation-related products, such as recombinant proteins, and others.
- Our customizable services are also designed to meet your specific needs, whether you are a researcher in academia or the biopharmaceutical industry.
- Apart from our products and services, we provide an abundance of resources covering various aspects of coagulation, comprising pathways, protein function, interacting proteins, articles, research areas, and other relevant topics.
Creative BioMart is dedicated to supporting your research and development undertakings in the domain of coagulation. Please feel free to explore our products, leverage our tailor-made services, and utilize our comprehensive resources to further your scientific pursuits.
If you have any questions, requirements, or cooperation intentions, please feel free to contact us. We very much look forward to working with you and helping you achieve research and commercial success.
References:
- Hill CN, Hernández-Cáceres MP, Asencio C, Torres B, Solis B, Owen GI. Deciphering the Role of the Coagulation Cascade and Autophagy in Cancer-Related Thrombosis and Metastasis. Front Oncol. 2020;10:605314.
- Palta S, Saroa R, Palta A. Overview of the coagulation system. Indian J Anaesth. 2014;58(5):515-523. doi:10.4103/0019-5049.144643