


Chromatin Immunoprecipitation (ChIP)
Chromatin Immunoprecipitation (ChIP) principle and introduction
Chromatin immunoprecipitation (ChIP) has become an important and popular method for the study of DNA-protein interactions. During the ChIP experiment, protein complexes that interact with DNA are cross-linked to their binding sites, and the chromatin is sheared into short fragments of 200-500 bps. After the DNA is sheared, the DNA fragments that interact with the protein of interest are isolated by immunoprecipitation. In traditional ChIP assays, the enriched immunoprecipitated DNA are quantified with quantitative PCRs using primer pairs designed to amplify specific regions of interest.
The strength of ChIP has been greatly increased by its combination with DNA microarray hybridization (ChIP-chip) to simultaneously analyze all putative binding sites displayed on the hybridization slide. In ChIP-chip, amplified immunoprecipitated DNA and total input DNA are labeled with distinct fluorescent dyes and co-hybridized to DNA microarrays containing probes.
The ChIP-chip method has many advantages over traditional ChIP assays. First of all, ChIP-chip microarray studies instead of interrogating a limited number of regions with specific primers, large genomic regions selected are probed in a single experiment, providing the potential discovery of unanticipated sites of protein-DNA interaction in a partially un-biased approach. A second advantage is that identification of protein binding sites can be accomplished with commercially available software platforms, permitting many parallel analyses of thousands of genes.
The strength of ChIP has been greatly increased by its combination with DNA microarray hybridization (ChIP-chip) to simultaneously analyze all putative binding sites displayed on the hybridization slide. In ChIP-chip, amplified immunoprecipitated DNA and total input DNA are labeled with distinct fluorescent dyes and co-hybridized to DNA microarrays containing probes.
The ChIP-chip method has many advantages over traditional ChIP assays. First of all, ChIP-chip microarray studies instead of interrogating a limited number of regions with specific primers, large genomic regions selected are probed in a single experiment, providing the potential discovery of unanticipated sites of protein-DNA interaction in a partially un-biased approach. A second advantage is that identification of protein binding sites can be accomplished with commercially available software platforms, permitting many parallel analyses of thousands of genes.
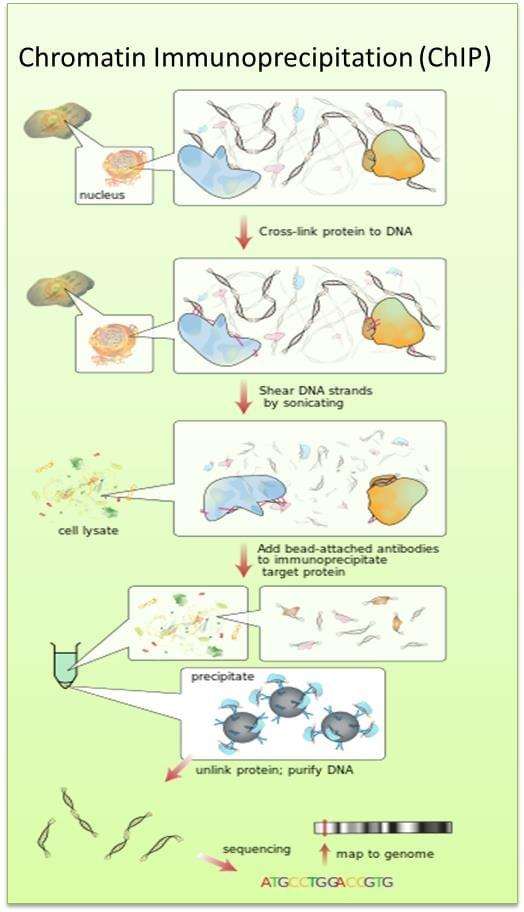
Fig. The schematic diagram of chromatin immunoprecipitation.
Chromatin immunoprecipitation (ChIP)
- Cells are fixed with 10% formaldehyde for 5 to 10 min.
- The crosslinking reaction is stopped by addition of glycine (0.125 M) for 5 min.
- Remove the medium and wash cells twice with 1′PBS.
- Scrape cells with cell lifter and pellet cells by centrifugation at 1500 rpm ′ 10 min at 4°C .
- Cells are hypotonically lysed in lysis buffer for 10 min on ice and homogenized.
- Collect nuclei by centrifugation at 1000 rpm ′ 10 min at 4°C.
- The nuclei are resuspended in 100 μl of Nuclear Lysis Buffer (50 mM TrisHCl pH7.6, 10 mM EDTA, 1% SDS) containing PI (B).
- Then the tube is washed with 100μl of IP Dilution Buffer (0.01% SDS, 1.1% Triton x100, 167 mM NaCl, 16.7 mM Tris-HCl pH7.6, 1.2 mM EDTA) containing PI (B) twice.
- The samples are sonicated for 10 minutes, with 10-second intervals.
- To check the sonication, 20 μl of chromatin are diluted to 100 μl with IP Dilution Buffer, added with 1 μl of RNase, NaCl to 0.3M for reverse crosslinking at 65°C for 6 hrs. The remaining chromatin samples are store at -80°C.
- The DNA from the aliquot that has the crosslinking reversed was purified using PCR purification columns.
- DNA is eluted in 30 μl and 4 μg of the DNA is loaded on 1% agarose gel for visualization of the fragment sizes.
- If DNA is sonicated sufficiently (200-300 bp), then the nuclear extract is diluted about 20-fold with IP Dilution Buffer + PI (B).
- DNA is equalized with Buffer B (0.05% SDS, 1% triton x100, 150 mM NaCl, 20 mMTris-HCl pH7.6, 2 mM EDTA) containing PI (B) by OD260 (0.3 to 0.6) of 1/20 dilution, by using 1/20 dilution of Buffer B as a blank.
- The chromatins are aliquoted 500 μl per tube and stored at -80°C.
- Sonication is repeated until fragment sizes were 200-300 bp.
- Prepare 200 μl of protein A/G agarose for each IP.
- The protein A/G agarose beads are washed with Buffer B three times by spinning down at 2000 rpm for 30 sec each time.
- The beads are blocked with 1mg/ml BSA fraction V and 667 μg/ml sonicated salmon sperm DNA on a rotator at 4°C for 1h or overnight. Each ChIP sample (500 μl) is added with PI (B) and spun down at 10,000 rpm ′ 10 minutes at 4°C, and the supernatant is transferred to new tubes.
- To each supernatant, 100 μl of blocked protein A/G beads is added and put on rotator at 4°C for 3h.
- The tubes are spun at 2000 rpm ′ 1 min and the flowthrough is saved for immunoprecipitation.
- Each antibody is added to flow-through as follows: 10 μl of FXR (20 μg total) and 2 μl of antibody (20 μg total). Samples are incubated on a rotator at 4°C overnight.
- 100 μl of blocked beads are added to each IP followed by incubation on a rotator for 2 hours at 4°C.
- After spinning at 2,000 rpm ′ 1 min, flow-through is removed and flow-through of antibody is saved as input DNA.
- Beads are washed with 500 μl of each buffer solution in the following order: Buffer B twice, Buffer D (0.05% SDS, 1% triton x100, 500 mM NaCl, 20 mM Tris-HCl pH7.6, 2 mM EDTA) twice, Buffer 3 (0.25 M LiCl, 1% NP40, 1% Deoxycholate, 10 mM Tris-HCl pH7.6, 1 mM EDTA) once, IP Wash Buffer (0.1% triton x100, 150 mM NaCl, 20 mM Tris-HCl pH7.6, 2 mM EDTA) once.
- To elute DNA-protein complex, 75 μl of IP Elution Buffer (1% SDS, 0.1M sodium bicarbonate) is added to the tube containing the beads and the tubes are vortexed for 10 minutes gently and spun down at 2,000 rpm x r 10 seconds.
- The elution process is repeated four times and the eluents are combined (300μl). The eluents are added with NaCl to 0.3 M and 1 μl of 10 mg/ml RNase, then reverse-crosslinked at 65°C for 4 hours and stored at -80°C.
- Purify DNA by using a PCR purification kit.
- The DNA will be analyzed by qPCR method.