


DNase I Footprinting
Principles
DNase I footprinting assay is an in vitro method to identify the specific site of DNA binding proteins. It not only finds the target protein that binds to specific DNA, but also identify which sequence the target protein is bound. This technique can be used to study interactions between proteins and DNA both outside and within cells.
The protein binds to the DNA fragment, protecting the binding site from cleavage by the DNase I. The fragments of the DNA molecule are left after cleavage (also known as the "footprinting", and thus its sequence can be determined. On the autoradiogram of the polyacrylamide electrophoresis gel, there is no radiolabeled band corresponding to the site of protein binding. Footprinting assay is specific, accurate positioning, and widely used.
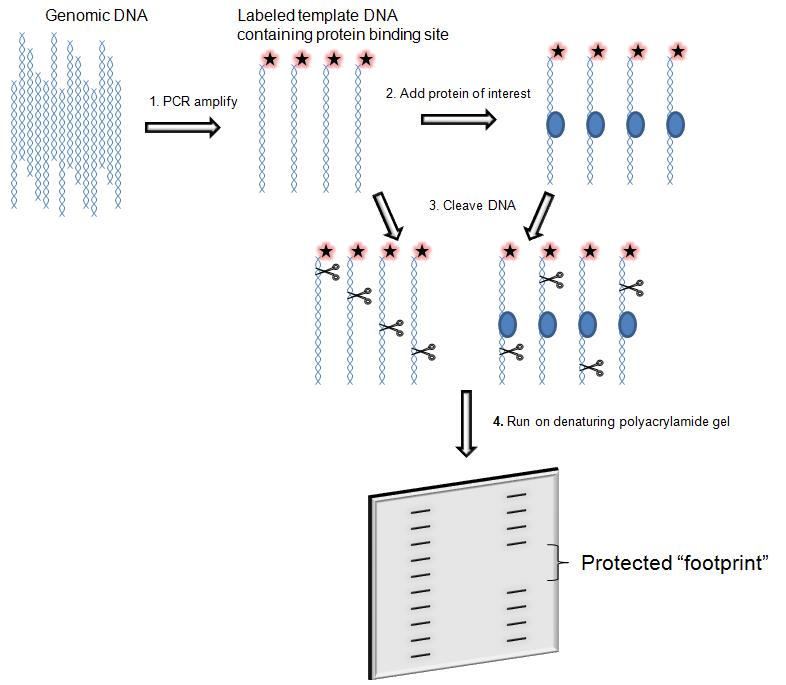
Regulation of transcription has been extensively studied, but there is still much that is not known. Transcription factors and related proteins that drive or repress transcription in conjunction with promoters, enhancers, or silencers are crucial to understanding the unique regulation of individual genes within the genome. Techniques like DNase I footprinting help clarify which proteins bind to these relevant regions of DNA and reveal the complexities of transcriptional control.
Advanced Applications
1. In vivo footprinting
In vivo footprinting combined with immunoprecipitation can be used to evaluate protein specificity at many positions throughout the genome. DNA binding to the protein of interest can be immunoprecipitated with an antibody to the protein, and then specific region binding can be assessed using the DNA footprinting technique.
2. Quantitative footprinting
The DNA footprinting technique can be modified to assess the binding strength of a protein to a DNA region. Using different concentrations of the protein for the footprinting experiment, the appearance of the footprint can be observed as the concentrations is increased and the proteins binding affinity can then be estimated.
3. Detection by capillary electrophoresis
To adapt the footprinting technique to newer detection methods, the labelled DNA fragments are detected by a capillary electrophoresis apparatus rather than run on a polyacrylamide gel. If the DNA fragment to be analyzed is generated by polymerase chain reaction (PCR), a fluorescent molecule such as carboxyfluorescein (FAM) is directly coupled to primers. This way, the fragments produced by DNase I digestion will contain FAM, and may be detected by capillary electrophoresis machine.
The following materials and reagents, as well as procedures are available for DNase I footprinting detected by polyacrylamide gel electrophoresis (PAGE).
Materials and Reagents
|
32P-labeled DNase I footprint probe Phenol / chloroform mixture (1: 1) Ethanol (100% and 75%) Polyvinyl alcohol (10%) Competition DNA Buffer Z' (or buffer Ze) |
DNase I (2.5mg/ml) MgCl2 (10mmol/L) CaCl2 (5mmol/L) DNase I stop solution Proteinase K (2.5 mg/ml) Formamide loading buffer |
Procedures
1. Bind Protein to DNA Probe.
1) Place a 1.5ml plastic microcentrifuge tube on ice to precool. Do not have to cover before adding DNase I stop solution.
2) The combination probe DNA mix for all reactions was formulated as follows (1X probe DNA mixture for one reaction):
| 32P-labeled DNase I footprint probe | Xμl |
| Polyvinyl alcohol (10%) | 10μl |
| Calf thymus DNA (1 mg/ml) | 0.2~1μl |
| H20 | Yμl |
| Total | 25μl |
Shake to thoroughly mix probe DNA mixture. Polyvinyl alcohol is viscous, only thoroughly shaken to allow complete mixing of the probe DNA solution.
Note: Competitive DNA may or may not be useful, but it is often beneficial to use competitive DNA in the analysis of crude protein fractions.
3) Add a suitable amount of buffer Z' and 0.1mol/L KCl to each reaction tube.
Note: The final volume of the buffer Z' and protein mix should be 25μl, and the final concentration of KCl in the mix should be 0.1mol/L.
4) The protein fraction is added directly to the reaction tube containing the appropriate amount of buffer Z'.
5) Add 25μl of probe DNA mix to each reaction tube, mix gently by flick and incubate on ice for 15 min.
2. Add the appropriate amount of DNase I to cleave DNA. The amount of DNase I is controlled so that only one phosphodiester bond cleavage occurs for each DNA. Set up protein-free controls.
6) Take a 2.5mg/ml DNase I stock solution to thaw. Dilute DNase I with ice-cold water, invert and gently shake to thoroughly mix.
Note: For a protein-free negative control reaction, 2μl of 1: 2000 diluted DNase I can be used for most DNA probes. The optimal amount of DNase I for a given DNA sequence may vary from sequence to sequence.
7) Remove a sample of DNA probes and protein fractions from ice and leave for 1 min at room temperature.
Note: It is helpful to have all the necessary equipment, such as automatic pipettes, solutions, timers, oscillators, and DNase I, present in an orderly fashion. The smoother this procedure will be, the better the footprint analysis will be (and the better the repeatability will be).
8) Add 50μl 10mmol/L MgCl2 and 5mmol/L CaCl2 solution (room temperature) in the reaction tube, flick mixed. Leave at room temperature for 1 min.
9) Dilution of DNase I was added to the reaction tube and immediately flick mixed. Incubate at room temperature for 1 min.
Note: Endogenous nucleases in the crude protein component alter the digestive ladder of DNase I. Therefore, in the footprinting analysis of crude extracts, a control reaction containing only the extract without DNase I should be included.
10) Add 90μl DNase I stop solution. Shock mixed.
Note: Typically, three (or more) samples are analyzed per cycle.
3. Remove the protein from the DNA, the modified DNA was loaded in the polyacrylamide electrophoresis gel and conducted autoradiography. The footprints of the nucleotide sequence are identified compared with the control group.
11) After all footprinting reactions have been completed, add 10μl of 2.5mg/ml proteinase K, mix gently and incubate at room temperature for 5 min. This step is useful for crude protein extracts, but not for pure protein. (This step is optional)
12) To prepare electrophoretic samples, add 200μl of 1: 1 phenol/chloroform solution, shock mixed. Centrifuge for 5 min.
13) Transfer the upper to a new tube, add 800μl 100% ethanol. Mix by inversion and centrifuge for 15 min to pellet the nucleic acid.
14) Remove the supernatant. Add 800μl 75% ethanol. Shock mixed. Centrifuge for 5 min.
15) Remove the supernatant. The precipitate was dried with a SpeedVac rotary concentrator.
16) Add 9μl formamide loading buffer and shake to dissolve the pellet.
17) Boil 3 min, set on ice to cool.
18) Centrifugation 0.5s to make solution sink to the bottom of the tube. Shock mixed. Add 4ul in the polyacrylamide electrophoresis gel.
Note: To determine the location of the protein binding site, a digested sample of DNase I can be added to the lane adjacent to the Maxam-Gilbert sequencing ladder (prepared from the footprint probe).
Example Data
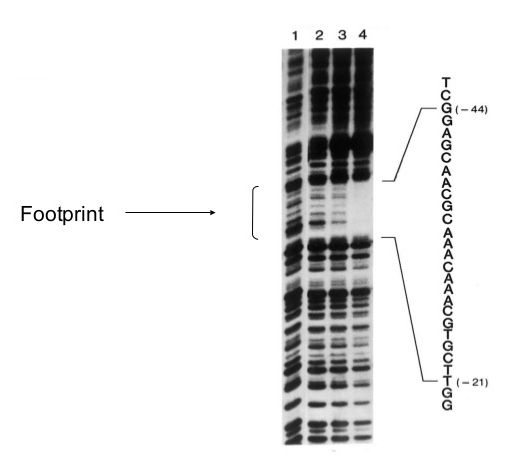
Samples in lanes 2-4 had increading amounts of the DNA binding protein (lambda protein cll); lane 1 had none.