


Methylation Interference Assay
Background and Principle
Many cell related life processes involve the interaction between DNA and proteins. With the development of recombinant DNA technology, a wide range of critical genes have been isolated. Now the key issue is to reveal how environmental factors and developmental signals controlling gene transcription. Therefore, it is necessary to identify and analyze DNA elements involved in the regulation of gene expression, and to isolate and identify these cis-element-specific protein factors. All these studies involve in the interaction between DNA and proteins.
Methylation interference assay is a method of identifying which regions of DNA strands bind to specific proteins by methylation modification of nucleotides. The rationale is that dimethyl sulfate (DMS) methylates the exposed guanine residue (G) in DNA, the piperidine in turn chemically cleaves methylated guanine residues. Methylation interference assay can be used to study the relationship between transcription factors and guanine residues in DNA binding sites. It is also an effective complement to DNase I footprinting assay to identify the precise location of DNA-protein interactions.
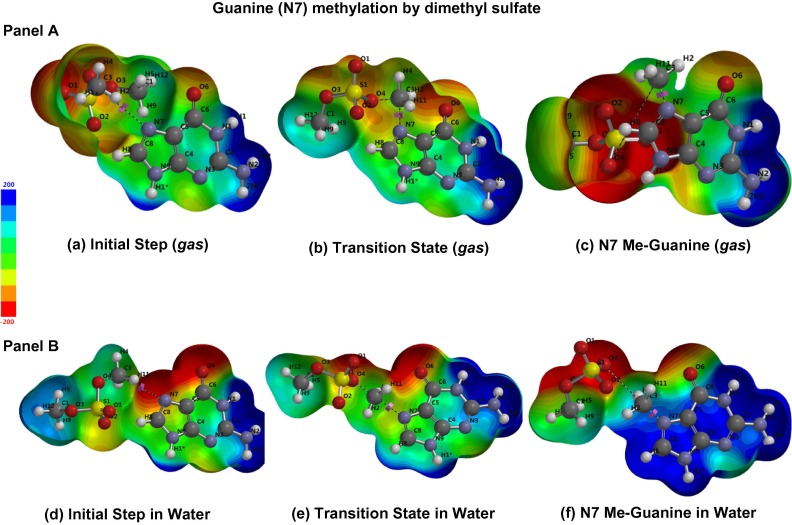 Figure 1. Guanine methylation by dimethyl sulfate. (Eichler D.R; et al. 2017)
Figure 1. Guanine methylation by dimethyl sulfate. (Eichler D.R; et al. 2017)
An advantage of methylation interference assay is the ability to detect the effect of the preferential methylation of guanine residues in the target DNA on protein binding, revealing in more detail the pattern of DNA-protein interactions. The disadvantage is that DMS can only methylate guanine and adenine residues in the DNA sequence while not methylate thymine and cytosine.
Procedure
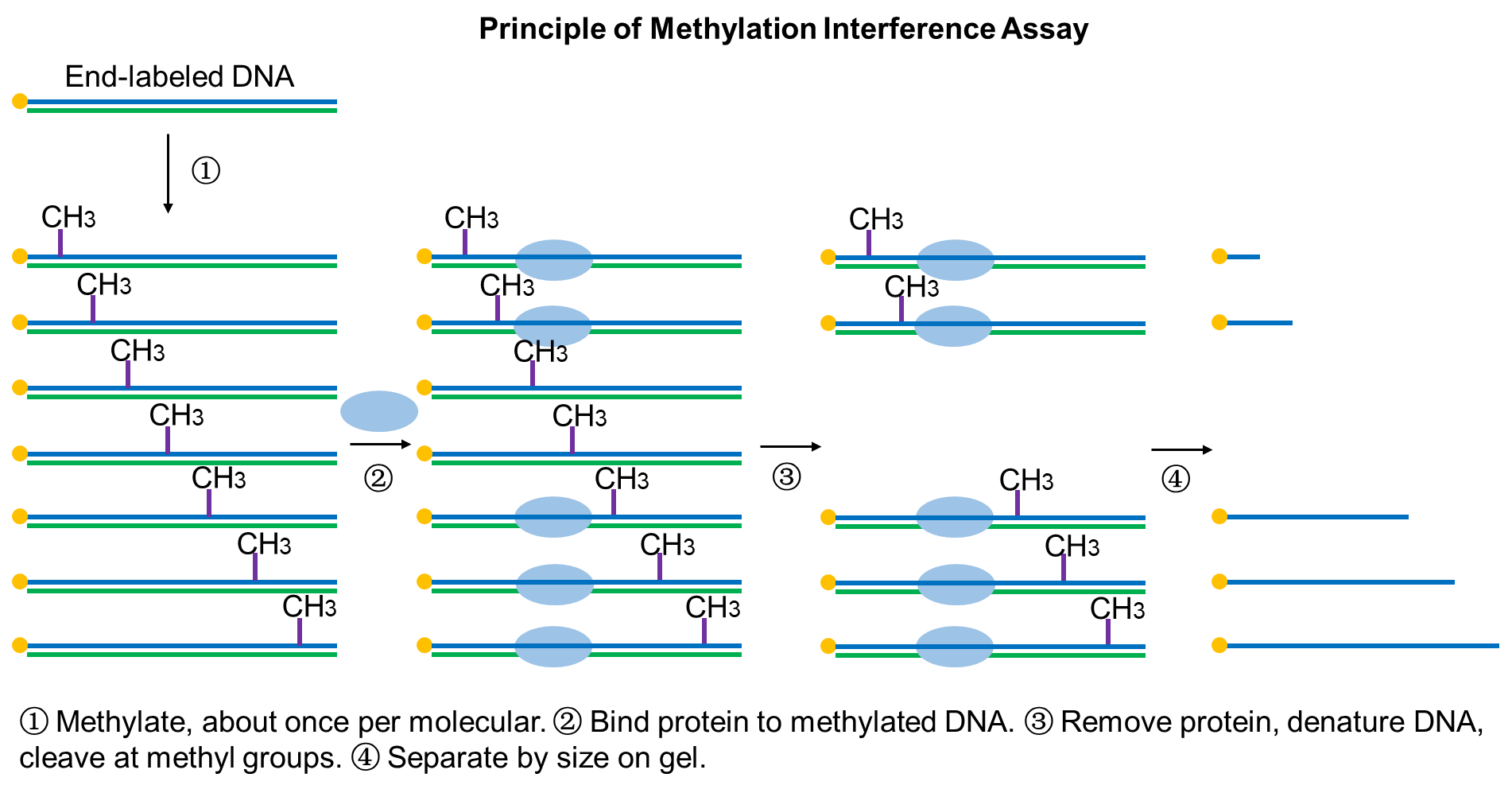
Ⅰ. Preparation of radiolabeled DNA probe
1. Preparation of polyacrylamide gel: 10 × TBE was diluted to a working concentration of 0.5 × TBE. The working concentration of polyacrylamide depends on the size of the DNA fragment to be tested. A gel of about 4 to 5% is required for a fragment of about 200bp, and a gel of 6 to 8% is prepared for a fragment of less than 100bp.
2. DNA labeling.
1) The DNA fragment to be tested was excised from 10μg of plasmid using two restriction enzymes. Be sure to have at least one enzyme digested to produce a 5' bulge. The final volume of the enzyme digestion system was 50μl and reacted for 1 hour under the appropriate enzyme reaction buffer conditions.
2) Add the following reagents to the enzyme reaction mixture. The reaction was incubated at room temperature for 30 minutes.
| [α-32P] dATP (10μCi/μl) | 10μl |
| dCTP (2mmol/L) | 1μl |
| dGTP (2mmol/L) | 1μl |
| dTTP (2mmol/L) | 1μl |
| DNA polymerase | 1μl |
3) Stop the reaction by adding 7μl of 10 × loading buffer.
3. Separation of labeled probe.
1) Load reaction mixture on non-denaturing gel.
2) Conduct electrophoresis 2 to 3 hours using 0.5 × TBE buffer under 10~15V/cm conditions.
3) Remove the glass plate from the cell, detach it and allow the gel to adhere to either plate. Cover the gel with plastic wrap.
4) The X-ray film is exposed to gel for one minute at room temperature. Careful operation allows the developed X-ray film and gel to recover from exposure.
5) Cover with cling film on radiographic images. The gel-coated glass plate was then placed thereon, using the photo as a template, and the gel slice containing the labeled DNA fragment was excised with a razor blade. The gel pieces were cut into small pieces (about 1mm) and placed in a microcentrifuge tube.
6) Add 1ml of elution buffer to the tube and shake at 37°C overnight. Incubate overnight.
7) Transfer the eluate into two clean microcentrifuge tubes (500μl per tube), then add 1 ml 100% ethanol each and keep -80°C for 10 minutes. The DNA is pelleted by centrifugation at room temperature for 15 minutes.
8) Discard the supernatant, wash the pellet with 80% ethanol, centrifuge for 5 minutes at room temperature, remove the supernatant with a long-necked Pasteur pipette and then dry the precipitate by vacuum centrifuging the concentrator or dryer.
9) Determine Cherenkov's radiation in the sample using a scintillation counter. The pellet was dissolved in an appropriate amount of TE so that the concentration of the labeled probe was about 104cpm/μl.
Ⅱ. Methylation reaction
The following reaction was established in a microcentrifuge tube at room temperature (6 tubes for each DNA fragment, 3 for the initial reaction and 3 for the termination reaction).
| Initial reaction | Tube 1: C reaction | Tube 2: G reaction | Tube 3: A+G reaction |
| DMS buffer | 200μl | 200μl | -- |
| H2O | -- | -- | 18μl |
| DNA Vector | 1μl | 1μl | 1μl |
| DNA probe | 1μl | 10μl | 2μl |
| Termination reaction | Tube 4: C termination | Tube 5: G termination | Tube 6: A+G termination |
| DMS stop solution | 50μl | 50μl | -- |
| Sodium acetate (0.3mol/L, pH 7.0) | -- | -- | 200μl |
| Ethanol (100%, ice precooled) | 1ml | 1ml | 1ml |
2. Add 1μl of DMS to tubes 1 and 2 and add 3μl of 10% formic acid to tube 3 to initiate the reaction.
3. Release tubes 1 and 2 for 4 minutes at 16°C and tube 3 for 15 minutes at 37°C. Add the appropriate amount of stop solution to terminate the reaction.
4. Vortex the tube vigorously and place in a dry ice/ethanol bath for 6 minutes.
5. Centrifuge at 4°C for 7 minutes.
6. Using a Pasteur pipette, discard the supernatant and wash the pellet with 1ml of 100% ethanol.
7. Centrifuge at room temperature for 3 minutes.
8. Discard the supernatant.
9. Drying the pellet with a desiccator or a vacuum centrifugal evaporator and concentrating the DNA pellet of tube 2 for the binding reaction, and storing tubes 1 and 3 at -20°C until use.
Ⅲ. Protein / DNA binding reaction
1. Preparation of polyacrylamide gel: 5% non-denatured polyacrylamide gel was poured into the gel. Final concentration of 1 × TGE was prepared with 10 × TGE.
2. Binding reaction.
1) Dissolve the dried, methylated, labeled DNA pellet (tube 2) with 23μl H2O.
2) Add the following reagents:
| poly(dI-dC) (1μg/μl) | 2μl |
| Protein extracts | 10~20μl |
| Binding buffer (10 ×) | 5μl |
| H2O | Xμl |
| Total | 50μl |
3) Perform binding reaction for 30 minutes at room temperature.
4) Add 3μl of loading buffer to the sample and load a 5% non-denaturing polyacrylamide gel.
5) Conduct electrophoresis 2 to 3 hours using 0.5 × TGE buffer under 10V/cm conditions to separate free and protein-bound DNA fragments.
6) Remove the glass plate from the cell and separate the two glass plates so that the glue attaches to a glass plate covered with a membrane.
7) Gel on the X-ray exposure of 1 to 2 hours at room temperature, be careful radiography self-imaging film imaging, so that the film coincides with the gel.
Ⅳ. Elution of radiolabeled DNA band
1. Wrap autoradiographic film with wrap and place on a disposable white tray. Unfold the gel, the gel-supporting glass plate is placed on the developed film such that the corners of the glass plate are aligned with the corners of the film, and the developed film is used as a template. The strips of polyacrylamide gel containing radioactively labeled DNA fragments (including free and protein-bound fragments) were excised with a knife.
2. Dissolve 1.5g agarose in 100ml of 1 × TBE, heat and melt in a microwave oven and cool to 60 ° C, pour the gel into 1.5% agarose gel and size 6.5cm × 10cm × 0.5cm microgel is enough to meet the experiment.
3. Cut a 0.5cm × 1cm hole on a piece of agarose gel with a scalpel and fill with 1 × TBE.
4. Place a 0.5cm × 0.5cm NA45 membrane in each well and ensure that the membrane extends to the anode.
5. Using a tweezers, place small piece of polyacrylamide gels in each well. The voltage gradient allows the DNA to migrate to the DNA45 membrane and run in 1 x TBE buffer. During the elution process, the power is turned off after DNA migration. The small gel is removed with tweezers.
6. Use tweezers to transfer the membrane into a microcentrifuge tube. Add 100 to 200μl of elution buffer to each tube to ensure that each membrane is completely immersed in the buffer.
7. Incubate at 68°C for 30-60 minutes.
8. The eluate was transferred to a new tube, add an equal volume of phenol/chloroform (1: 1), shaken vigorously.
9. Centrifuge at room temperature for 5 minutes and transfer the supernatant to a new tube.
10. Add 5μg of sonicated salmon sperm DNA to each tube, followed by 2 volumes of 100% ethanol and keep at -80°C for 10 minutes to precipitate DNA.
11. Collect the DNA by centrifugation at 4°C for 15 minutes.
12. Aspirate the supernatant with a Pasteur pipette and use a Geiger counter to detect the labeled DNA.
13. Wash the pellet with 500μl of 80% ethanol, shake, and centrifuge at 4°C for 5 minutes.
14. Discard the supernatant and dry the pellet with a desiccator or a vacuum centrifugal evaporator.
Ⅴ. DNA fragmentation and denaturing PAGE analysis
1. Preparation of denatured polyacrylamide gel (for sequencing).
1) Clean and silicidize two glass plates (33cm x 42cm and 33cm x 39cm) and rub the surface lightly with 100% ethanol. The cells of the two glass plates are separated by a 0.2mm thick pad to assemble the glass plate.
2) Add 200μl 10% APS and 200μl TEMED to 100ml denatured adhesive solution, pour into the prepared glass immediately after mixing, insert the comb, and allow the glue to polymerize for 1~2 hours.
3) Remove the comb and place the glass plate in the electrophoresis tank. Add 1 × TBE to the upper and lower tanks, rinse the wells with 1 × TBE with a Pasteur pipette, and pre-electrophoresis for 1 hour at 40~50W. It is important to preheat the glue before loading.
2. Cutting reaction
1) Add 100μl of 10% piperidine in each dried DNA pellet.
2) Incubate at 90°C for 30 minutes.
3) Use a needle to puncture a small hole in each tube cover, lyophilize the cleaved DNA sample in a vacuum centrifuge evaporator concentrator for 2 to 5 hours or until completely dry.
4) Determination of Cherenkov dose per DNA sample by scintillation counting.
5) Add the sequencing stain to the DNA sample so that the final sample concentration for sample 1~3 is 5000cpm/μl and the sample 4 should be 104cpm/μl.
6)Denature DNA by treating at 100°C for 10 minutes, then cool on ice.
7)Load the modified polyacrylamide gel pre-heated at 40-50 W for 1 hour in the loading wells, 5μl on each sample.
8)According to the length of DNA fragments, electrophoresis at 40~50W for 2~3 hours.
9)Remove the glass plates from the cell, attach the gel to one of the plates. Carefully place a piece of Whatman 3MM on a piece of glue, slowly peel the plate and cover the gel with a membrane and dried at 80 ° C for 1 hour. The dried gel was X-rayed at -80°C overnight to reveal the DNA band.
Result Analysis
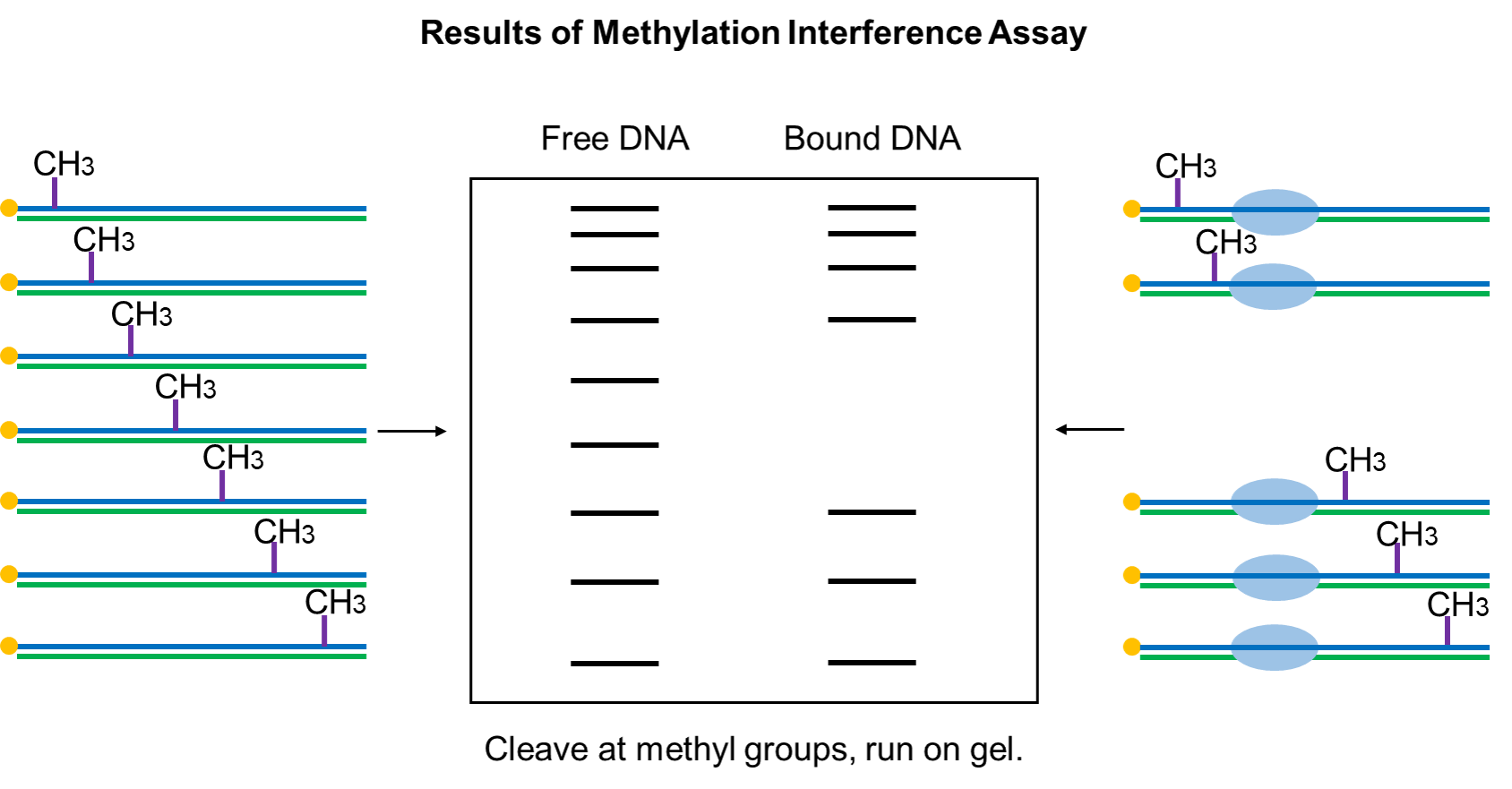
The target DNA sequence that binds to the transcription factor protein is cleaved by the piperidine, electrophoresis separation showed bands with a blank area. The target DNA sequence that does not bind to the transcription factor protein is cleaved by the piperidine, electrophoresis separation showed all bands, no blank area appears.
Reference
1. Eichler D.R; et al. Activation Barriers for Methylation of DNA Bases by Dimethyl Sulfate [J]. Chemical Physics Letters, 2017, 689.