
Hedgehog Family
Related Symbol Search List
Immunology Background
Background
The Hedgehog (Hh) family consists of a group of signaling proteins that play crucial roles in embryonic development, particularly in the regulation of cellular processes such as axon guidance and neuron migration. The three main members of this family are Sonic Hedgehog (Shh), Indian Hedgehog (Ihh), and Desert Hedgehog (Dhh). These proteins are known for their effects on cell proliferation, differentiation, and patterning.
Hedgehog Signaling Pathway
The Hedgehog signaling pathway is essential for various developmental processes. It involves the binding of Hedgehog proteins to the Patched (Ptch) receptor, which in turn activates the Smoothened (Smo) protein, leading to transcriptional activation of target genes via the Gli family of transcription factors. This pathway is highly conserved across species and has been implicated in numerous biological processes, including:
- Neuronal Development: Critical for the proper development of the nervous system.
- Patterning of Tissues: Plays a role in the spatial arrangement of different cell types during development.
Axon Guidance
Axon guidance is the process through which growing axons navigate through the embryo to reach their appropriate targets. Hedgehog proteins contribute to this process by:
- Creating Gradients: Shh, for example, establishes a gradient that influences the direction of growth cone movement.
- Modulating Cell Adhesion: Hedgehog signaling can regulate the expression of adhesion molecules, affecting how axons interact with their environment.
Neuron Migration
Neuron migration is essential for the formation of brain architecture. During development:
- Radial Migration: Neurons migrate from the ventricular zone towards the cortical plate, guided by various signaling cues, including Hedgehog proteins.
- Tangential Migration: Involves longer-distance movement, where Hedgehog signaling can help direct these cells along specific paths.
Mechanisms of Action
- Hedgehog proteins exert their effects through a signaling pathway that involves interactions with Patched receptors and Smoothened transmembrane proteins, ultimately leading to the modulation of transcription factors like the Gli family.
- The activation of downstream target genes by Hedgehog signaling influences various cellular processes critical for axon guidance, neuron migration, and overall nervous system development.
The Hedgehog family of proteins plays a multifaceted role in orchestrating the intricate processes of axon guidance and neuron migration during neural development. Understanding the mechanisms by which Hedgehog signaling regulates these processes is essential for unraveling the complexities of nervous system formation and function.
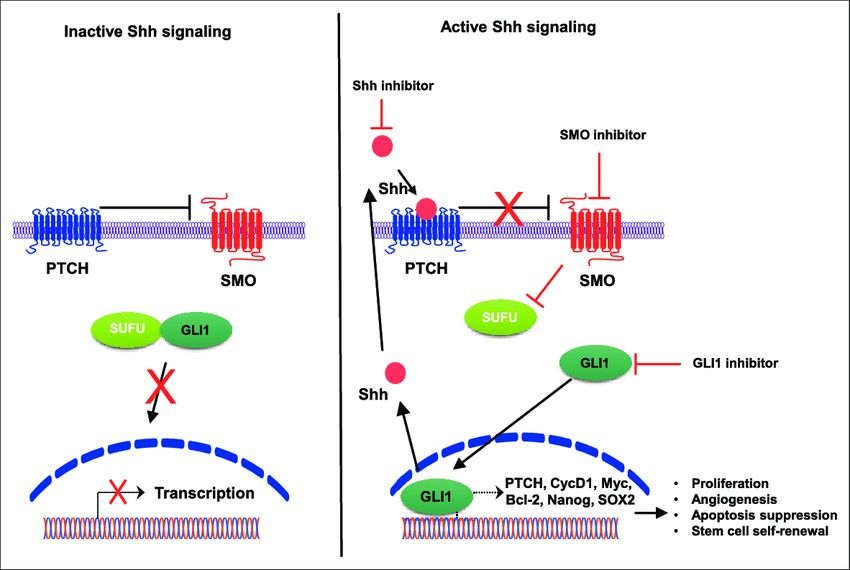 Fig.1 Hedgehog signaling pathway. (Lee DH, et al., 2017)
Fig.1 Hedgehog signaling pathway. (Lee DH, et al., 2017)Inactivated signaling (left) occurs in the absence of Shh ligands, wherein PTCH inhibits SMO resulting in GLI1 sequestration in the cytoplasm by SuFu. In the presence of Shh (right), PTCH suppression of SMO is abrogated, resulting in the nuclear accumulation of GLI1 and activation of target genes that promote several oncogenic properties in tumor cells.
Hedgehog Signaling Pathway-Related Molecules
Below is a brief overview of the many genes and proteins associated with the Hedgehog signaling pathway:
| Genes and Proteins | Details |
|---|---|
| BTRC (Beta-Transducin Repeat Containing E3 Ubiquitin Protein Ligase) |
|
| C2CD3 (C2 Calcium-Dependent Domain Containing 3) |
|
| CDO1 (Cell Adhesion Molecule-Related Down-Regulated by Oncogenes 1) |
|
| DISP2 (Dispatched RND Transporter Family Member 2) |
|
| GPR161 (G Protein-Coupled Receptor 161) |
|
| PTCH1 (Patched 1) |
|
These molecules play critical roles in regulating various aspects of the Hedgehog signaling pathway, contributing to its proper functioning in processes such as embryonic development, cell differentiation, and tissue patterning.
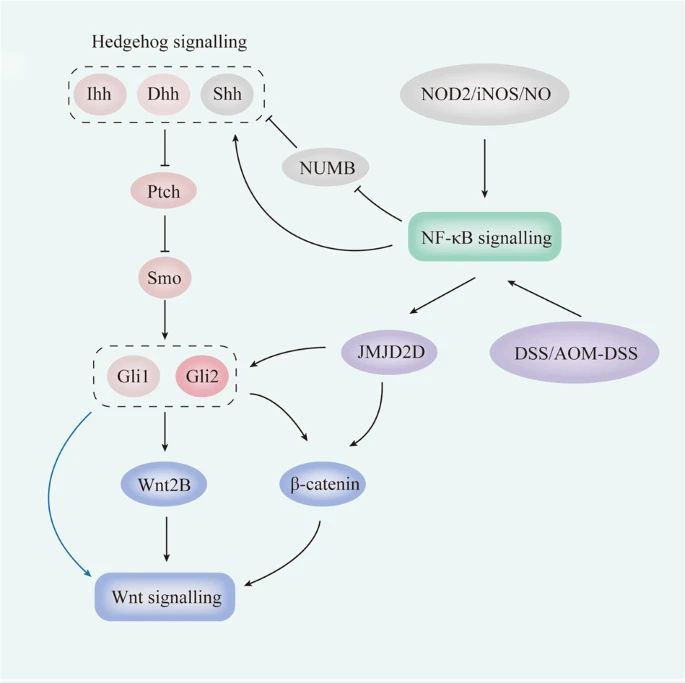 Fig.2 Crosstalk between Hedgehog and other pathways in the intestinal tract. (Xie Z, et al., 2021)
Fig.2 Crosstalk between Hedgehog and other pathways in the intestinal tract. (Xie Z, et al., 2021)Potential Implications of Hedgehog Signaling Dysregulation in Neurodevelopmental Disorders
Hedgehog (Hh) signaling is a critical pathway in embryonic development and tissue homeostasis, playing significant roles in neurodevelopment. Dysregulation of this pathway can have profound implications for neurodevelopmental disorders. Here are some potential implications:
Neuronal Differentiation and Proliferation
- Impaired Neuronal Development: Abnormal Hh signaling can lead to disrupted neuronal differentiation, potentially resulting in neurodevelopmental disorders such as autism spectrum disorder (ASD) and schizophrenia.
- Altered Cell Proliferation: Dysregulation can affect the proliferation of neural progenitor cells, potentially leading to developmental abnormalities or reduced neuronal populations.
Axon Guidance and Synaptogenesis
- Deficient Axon Guidance: Hedgehog signaling influences pathways involved in axon growth and guidance. Disruption could lead to misrouting of axons, contributing to cognitive and behavioral deficits.
- Synaptic Formation Issues: Hh signaling is implicated in synapse formation, and dysregulation could lead to altered synaptic connectivity, affecting learning and memory.
Regional Brain Patterning
- Abnormal Brain Structure: Hedgehog signaling plays a role in regional patterning of the brain during development. Dysregulation may lead to structural abnormalities, impacting function and behavior.
- Lateralization Effects: Disruption in Hh signaling could influence lateralization of brain functions, potentially resulting in dissociative cognitive processes.
Interaction with Other Pathways
- Wnt and Notch Pathways: Hh signaling interacts with other key developmental pathways. Dysregulation could lead to complex alterations in signaling networks, contributing to a variety of neurodevelopmental disorders.
- Inflammatory Responses: Abnormal Hh signaling may influence inflammatory pathways in the brain, potentially leading to neuroinflammation associated with conditions like ASD or ADHD.
Genetic and Environmental Interactions
- Genetic Predisposition: Mutations in Hh signaling components (e.g., SHH, SMO) can be linked to genetic neurodevelopmental disorders, suggesting a direct pathway from genetics to neurodevelopmental outcomes.
- Environmental Factors: Exposure to teratogens that affect Hh signaling during critical periods can lead to neurodevelopmental outcomes, highlighting the importance of this pathway in response to environmental stressors.
Long-Term Effects and Behavior
- Behavioral Abnormalities: Dysregulated Hh signaling may lead to long-term behavioral problems such as anxiety, depression, or hyperactivity.
- Impact on Cognitive Functions: Changes in neuronal connectivity and synaptic function can ultimately affect cognitive abilities, leading to learning disabilities or intellectual disability.
In summary, dysregulation of Hedgehog signaling can have wide-ranging effects on neurodevelopment, potentially contributing to the pathophysiology of various neurodevelopmental disorders. Further research in this area may provide insights into therapeutic targets and interventions.
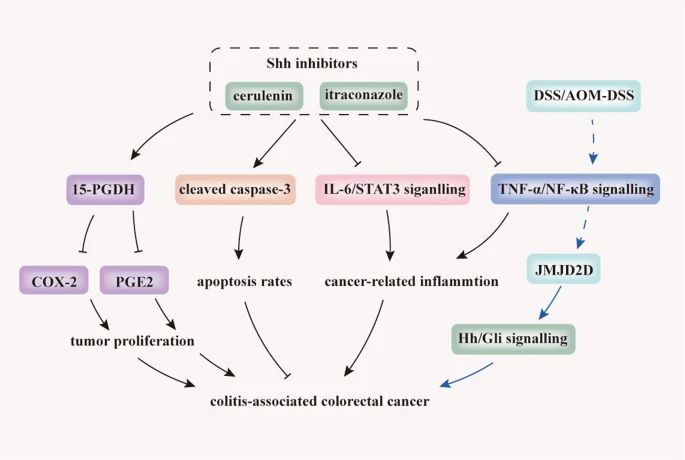 Fig.3 Inhibition of Hh signaling prevents tumourigenesis and CAC progression. (Xie Z, et al., 2021)
Fig.3 Inhibition of Hh signaling prevents tumourigenesis and CAC progression. (Xie Z, et al., 2021)Case Study
Case 1: Choi KS, Harfe BD. Hedgehog signaling is required for formation of the notochord sheath and patterning of nuclei pulposi within the intervertebral discs. Proc Natl Acad Sci U S A. 2011;108(23):9484-9489.
During mouse embryogenesis, the transient rod-like structure known as the vertebral notochord secretes factors that play a crucial role in organizing neighboring tissues. While the notochord typically develops into the nucleus pulposus within the intervertebral disc, the specific molecular processes involved in this transformation remain poorly understood. Recent research has revealed that the activation of hedgehog signaling is vital for the proper development of intervertebral discs.
When hedgehog signaling was experimentally disrupted in the notochord and adjacent floorplate, an abnormal notochord sheath formed around the structure, deviating from its usual configuration. This alteration led to the formation of smaller nuclei pulposi, with many notochord cells dispersing throughout the vertebral bodies as embryonic development progressed. These findings suggest that hedgehog signaling is essential for the formation of the notochord sheath, which in turn plays a critical role in maintaining the structural integrity of the notochord during early embryonic stages. As notochord cells transition into nuclei pulposi, it is proposed that the notochord sheath acts as a protective envelope, confining these cells along the length of the vertebral column.
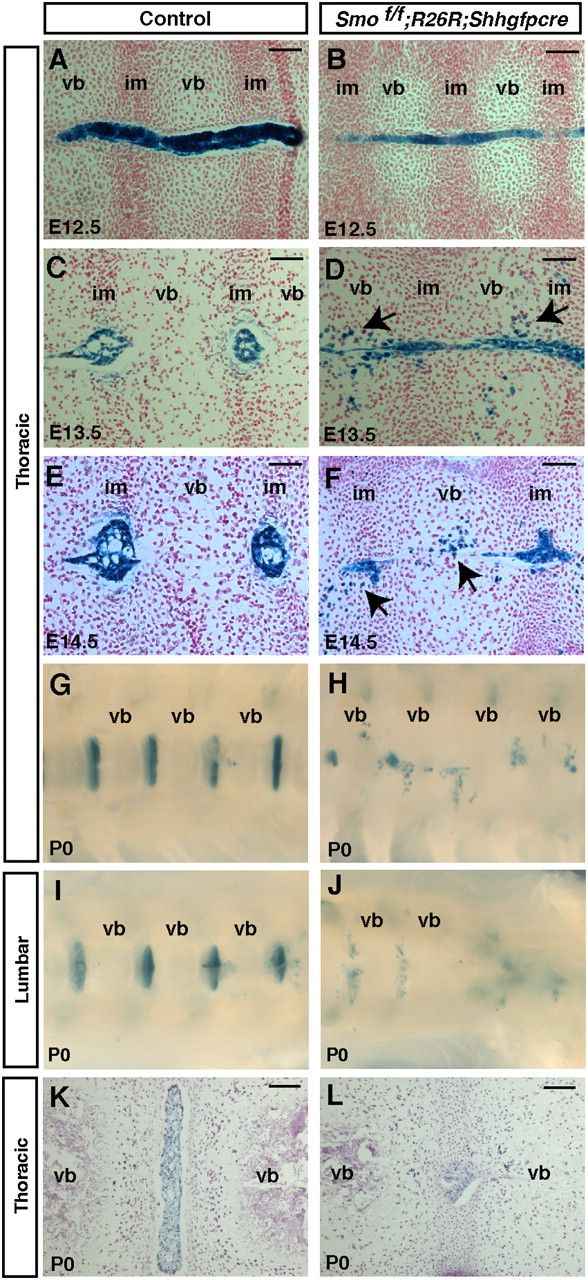 Fig.1 Aberrant migration of notochord cells throughout the vertebral column upon removal of hedgehog signaling in the notochord.
Fig.1 Aberrant migration of notochord cells throughout the vertebral column upon removal of hedgehog signaling in the notochord.Case 2: Hamilton AM, Balashova OA, Borodinsky LN. Non-canonical Hedgehog signaling regulates spinal cord and muscle regeneration in Xenopus laevis larvae. Elife. 2021;10:e61804.
In the realm of modern medicine, prompting regeneration in damaged spinal cords stands out as a significant challenge. Various studies across different model organisms suggest that Hedgehog (Hh) signaling could serve as a promising target for stimulating regeneration. However, the precise mechanisms through which Hh signaling facilitates tissue regeneration remain ambiguous.
In a study focusing on post-amputation tail regeneration in Xenopus laevis larvae, researchers delved into the role of Hh signaling. Their investigation revealed that while the activity of Smoothened (Smo) is crucial for the proper regeneration of the spinal cord and skeletal muscle, the transcriptional activity of the canonical Hh effector Gli is initially suppressed following amputation. Interestingly, inhibiting the expression or transcriptional activity of Gli1/2 showed limited impacts on the regenerative process.
On the contrary, the study demonstrated the vital role of protein kinase A in the regeneration of both muscle and spinal cord tissues. This kinase operates in conjunction with Smo for muscle regeneration and independently for spinal cord regeneration. Furthermore, the downstream effector of protein kinase A, CREB, was found to be activated in the spinal cord post-amputation in a Smo-dependent manner. These results suggest that non-canonical mechanisms of Hh signaling play a crucial role in the regeneration of spinal cord and muscle tissues.
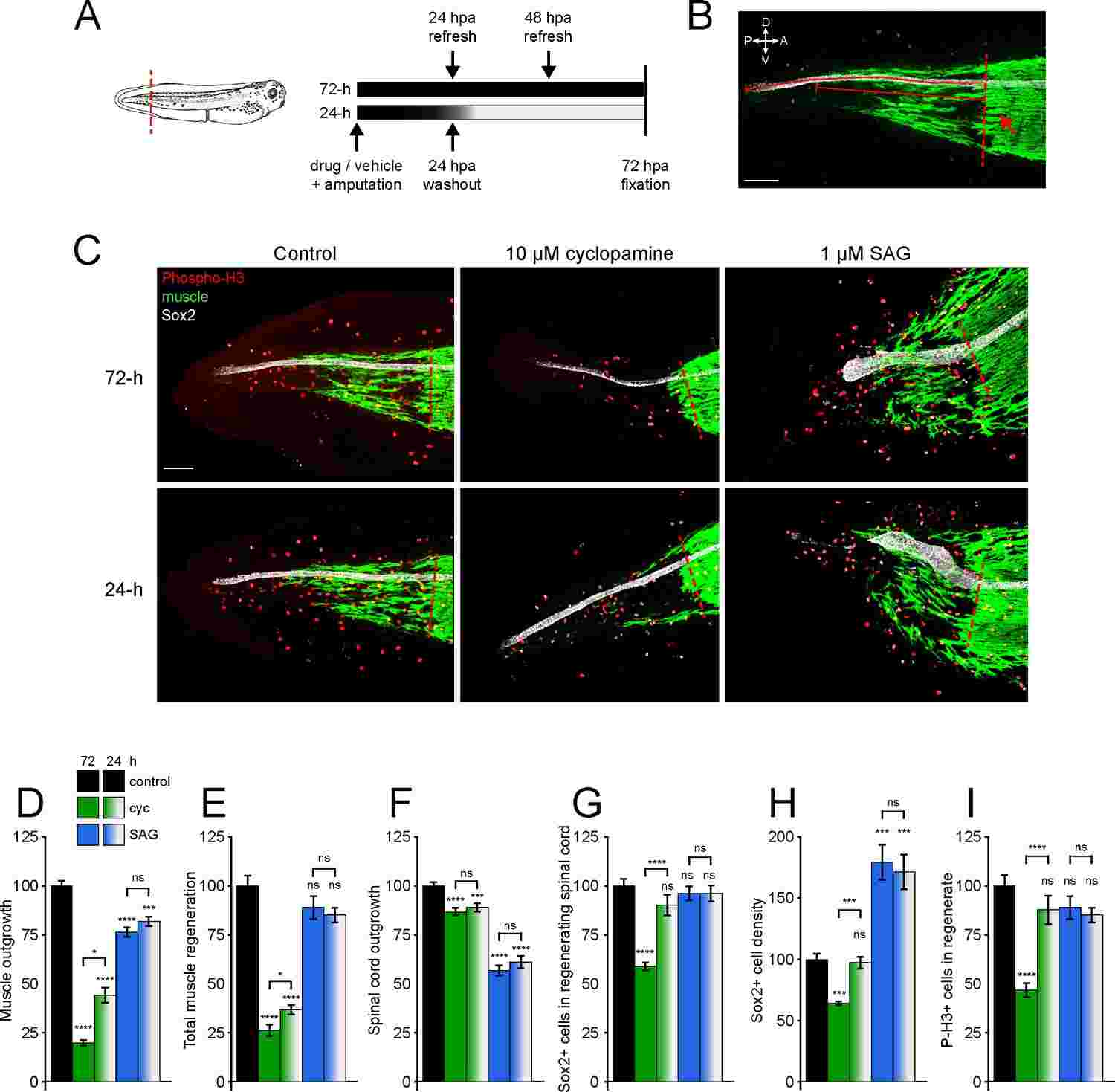 Fig.2 Hedgehog signaling regulates spinal cord and muscle regeneration.
Fig.2 Hedgehog signaling regulates spinal cord and muscle regeneration.Related References
- Lee DH, Lee SY, Oh SC. Hedgehog signaling pathway as a potential target in the treatment of advanced gastric cancer. Tumour Biol. 2017;39(6):1010428317692266.
- Xie Z, Zhang M, Zhou G, et al. Emerging roles of the Hedgehog signaling pathway in inflammatory bowel disease. Cell Death Discov. 2021;7(1):314.