
Principle and Protocol of Co-IP
Immunoprecipitation (co-immunoprecipitation) is a classical method for studying protein interactions based on the specific binding of antigen and antibody. Since many mutually bound proteins remain bound during cell lysis, immunoprecipitation of the target protein by specific antibodies will precipitate the proteins that interact with the target protein as well, and detect the interacting proteins by immunoblotting.
Immunoprecipitation is closer to the detection of protein-protein interactions in the intracellular physiological state, and is an effective method for determining intracellular physiological interactions. Therefore, interacting proteins screened by methods such as yeast two-hybridization often need to be tested for physiological interactions using immunoprecipitation.
The laboratory manual is designed to help researchers master the principles of immunoprecipitation and its applications, learn to apply immunoprecipitation methods to detect protein interactions, and become familiar with the operational procedures and precautions for immunoprecipitation techniques.
Immunoprecipitation is a method for studying protein-protein interactions based on the specific binding of antigens and antibody. Protein A/G is able to bind specifically to FC fragments of immunoglobulins. By pre-binding protein A/G to agarose (agarose) and reacting it with a solution containing antigen and antibody, protein A/G on agarose is able to precipitate antigen and antigen-bound protein by binding antibody and thereby. When cells are lysed under nondenaturing conditions, many of the proteins present in intact cells and the binding between proteins are preserved, and protein X is immunoprecipitated by antibodies to covalently bound protein A. If protein Y is bound to protein X in vivo, then it may also be precipitated, and a band of protein Y can be detected by immunoblotting (Figure 4-3-1).
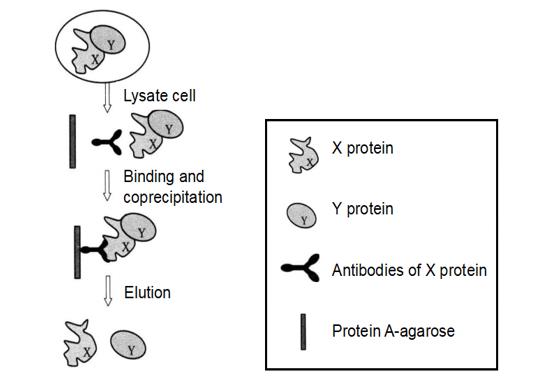 Figure 4-3-1 Schematic Diagram of the Principle of Immunoprecipitation
Figure 4-3-1 Schematic Diagram of the Principle of Immunoprecipitation
1. Main Instruments and Equipment
Micropipette (2 μL, 10 μL, 20 μL, 200 μL, 1000μL), low-temperature centrifuge, desktop centrifuge, agarose gel electrophoresis system, protein gel electrophoresis system, gel imaging system, semi-dry transfer system, thermostatic shaker, thermostatic incubator, fume hood, ice maker, oscillator, thermostatic metal bath.
2. Main Reagents
1. Protein extraction buffer/ wash buffer 1
| Tris-HCl (pH 7.5) | 50 mmol/L |
| NaCl | 100 mmol/L |
| MgCl2 | 4 mmol/L |
| EDTA | 2 mmol/L |
| NP-40 | 0.5% (Volume fraction) |
| DTT | 5 mmol/L |
| PMSF | 1 mmol/L |
| PMSF add before use |
2. Wash buffer 2
| Tris-HCl (pH 7.5) | 25 mmol/L |
| NaCl | 500 mmol/L |
| NP-40 | 0.1% (Volume fraction) |
| Sodium deoxycholate | 0.05% |
3. Wash buffer 3
| Tris-HCl (pH 7.5) | 25 mmol/L |
| NP-40 | 0.1% (Volume fraction) |
| Sodium deoxycholate | 0.05% |
4. Protein A-agarose (from Roche)
1. Extraction of Plant Tissue Samples
(1) In a 1.5 mL centrifuge tube, add 500 μL of pre-cooled protein extraction buffer with a small amount of PVP.
(2) Take 0.2g of plant tissue material into a pre-chilled mortar, pour in liquid nitrogen, grind it into powder form, and add it to a 1.5mL centrifuge tube containing protein extraction buffer.
(3) Shake vigorously and shake on ice for 10 min.
(4) Centrifuge at 12,000g, 4°C for 15min, and transfer the supernatant to a new 1.5mL centrifuge tube.
(5) Centrifuge at 12000g, 4°C for 15min, and collect the supernatant as total protein sample*1.
2. Immunoprecipitation
(1) Take 1 mL of tissue protein sample and add to a 1.5 mL centrifuge tube*2.
(2) Centrifuge at 12000g, 4°C for 10 min, transfer the supernatant to a new 1.5mL centrifuge tube and discard the precipitate.
(3) Add 50μL of protein A-agarose to the sample and incubate at least 3 h (or overnight) at 2 to 8°C with shaking*3.
(4) Precipitate agarose by gravity sedimentation or 12000g, centrifugation for 20 s. Transfer the supernatant to a new microcentrifuge tube.
(5) To the tissue protein sample, add 50μL of protein-specific antibody and incubate at 2 to 8°C, with shaking*4.
(6) Add 50μL of protein A-agarose to the sample and incubate at 4°C, shaking for at least 3h (or overnight).
(7) Collect agarose-antibody-antigen complexes by gravity sedimentation or 12000g, centrifugation for 20s.
(8) Carefully remove the supernatant and resuspend the agarose with 1 mL wash buffer 1 at 4°C and incubate with shaking for 20 min.
(9) Collect the agarose-antibody-antigen complexes by gravity sedimentation or centrifugation at 12,000g for 20s.
(10) Carefully remove the supernatant and resuspend the agarose with 1 mL wash buffer 2 and incubate for 20 min at 4°C with shaking.
(11) Collect the agarose-antibody-antigen complexes by gravity sedimentation or centrifugation at 12000g for 20 s.
(12) Carefully remove the supernatant and resuspend the agarose with 1 mL wash buffer 3 and incubate for 20 min at 4°C with shaking.
(13) Collect the agarose-antibody-antigen complexes by gravity sedimentation or centrifugation at 12,000g for 20s.
(14) Resuspend the agarose precipitate*5 with 50μL protein loading buffer.
(15) Heat at 100°C for 5 min to denature the protein.
(16) Centrifuge at 12000g for 20s at 15-25°C to remove protein A-agarose and transfer the supernatant to a new centrifuge tube.
(17) Half of the supernatant was taken for SDS polyacrylamide gel electrophoresis analysis.
(18) Detect whether Y protein interacts with X protein by immunoblotting.
1. If the binding of the two proteins is too weak and the sensitivity of immunoprecipitation is limited, FRET or yeast two-hybrid and other in vivo binding detection methods with higher sensitivity can be considered.
2. The combination of the two proteins requires a specific stimulus and can occur under specific circumstances. The stimulation and binding conditions that may be required for the binding of the two proteins can be found through functional research or literature review. The growth rate of cells affects the expression of foreign proteins, so it is necessary to optimize the state of bacterial growth before induction, the concentration of IPTG induction, the induction temperature and the induction time. The rapid growth of E. coli and the overexpression of foreign proteins easily lead to the formation of inclusion bodies, which is not conducive to the purification of proteins.
*1 The concentration of protein should be 5-20mg/mL.
*2 One immunoprecipitation reaction requires 1-3mL of sample. Only 1mL is needed for immunoprecipitation reaction in 1.5mL centrifuge tube.
*3 Remove the background caused by the binding of non-specific protein to protein A-agarose.
*4 The amount of X protein antibody should be determined according to the concentration of the antibody.
*5 The loading buffer should be added according to the amount of precipitation.