
B Cell Development and Differentiation
Creative BioMart B Cell Development and Differentiation Product List
Immunology Background
About Osteoblast and Osteoclast Markers
Osteoblasts and osteoclasts are two types of cells that play crucial roles in bone metabolism and remodeling. Osteoblasts are responsible for bone formation, while osteoclasts are involved in bone resorption. To identify and characterize these cells, researchers utilize specific markers that are expressed on their surface or in their cytoplasm. These markers help distinguish osteoblasts and osteoclasts from other cell types and provide insights into their functions and activity.
Here are some commonly used markers for osteoblasts and osteoclasts:
Osteoblast Markers
Alkaline Phosphatase (ALP): ALP is an enzyme expressed on the surface of osteoblasts during active bone formation. It plays a critical role in mineralization and is widely used as a marker for early osteoblast differentiation.
Osteocalcin (OCN): OCN is a non-collagenous protein secreted by mature osteoblasts. It is involved in regulating bone mineralization and is commonly used as a late-stage marker for osteoblast differentiation.
Osterix (OSX): OSX is a transcription factor essential for osteoblast differentiation and function. It is expressed in preosteoblasts and mature osteoblasts and is considered a key regulator of osteoblast-specific gene expression.
Runt-related transcription factor 2 (RUNX2): RUNX2 is a master regulator of osteoblast differentiation and bone development. It is expressed in preosteoblasts and plays a crucial role in the commitment of mesenchymal cells to the osteoblast lineage.
Collagen Type I (COL1A1): Collagen Type I is the main component of the organic matrix in bone. It is synthesized and secreted by osteoblasts during bone formation. Immunostaining for COL1A1 is commonly used to identify osteoblasts and assess their activity.
Osteoclast Markers
Tartrate-Resistant Acid Phosphatase (TRAP): TRAP is an enzyme highly expressed in osteoclasts and is involved in bone resorption. It is commonly used as a marker to identify and visualize osteoclasts in bone tissue sections.
Cathepsin K: Cathepsin K is a protease predominantly expressed in osteoclasts. It plays a crucial role in bone resorption by degrading bone matrix proteins. Cathepsin K staining is widely used to identify osteoclasts and assess their activity.
Receptor Activator of Nuclear Factor κB (RANK): RANK is a cell surface receptor expressed on osteoclast precursors and mature osteoclasts. The binding of RANK with its ligand, RANKL, is essential for osteoclast differentiation and activation.
Calcitonin Receptor (CTR): CTR is a G-protein-coupled receptor expressed on osteoclasts. It binds to calcitonin and plays a role in regulating osteoclast activity and bone resorption.
Matrix Metalloproteinase-9 (MMP-9): MMP-9 is an enzyme involved in extracellular matrix degradation and is expressed by osteoclasts during bone resorption. It is used as a marker for identifying active osteoclasts.
These markers assist researchers in identifying, characterizing, and studying osteoblasts and osteoclasts in various physiological and pathological conditions. They provide valuable tools for investigating bone metabolism, bone remodeling, skeletal development, and diseases such as osteoporosis, arthritis, and bone tumors. Understanding the functions and regulation of osteoblasts and osteoclasts is essential for developing therapeutic strategies targeting bone disorders and promoting bone health.
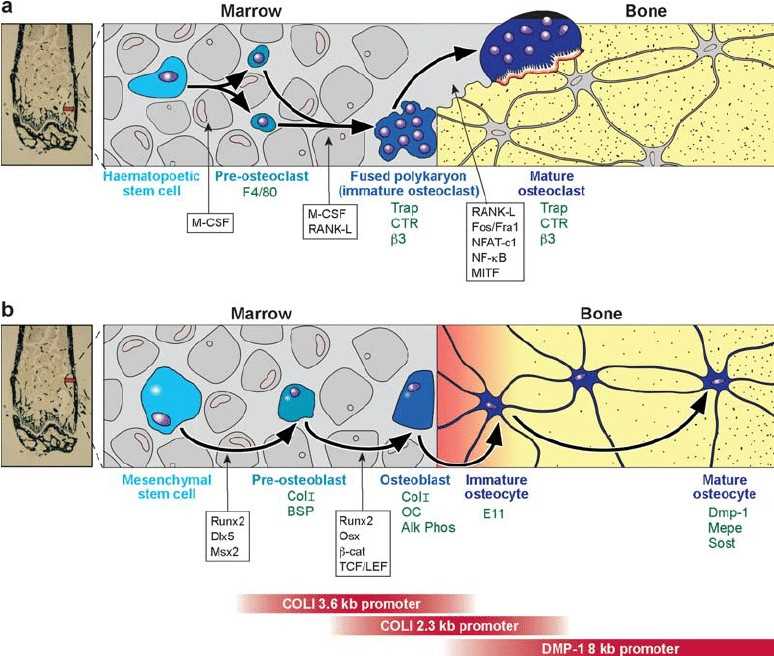 Fig. 1 Lineage of osteoclasts and osteoblasts. (Alexander G Robling, et al., 2006)
Fig. 1 Lineage of osteoclasts and osteoblasts. (Alexander G Robling, et al., 2006)
( a ) Osteoclasts are derived from a hematopoeitic precursor in the bone marrow, spleen, or liver. Proliferation of mononuclear cells from the precursor population requires M-CSF. The blood-borne preosteoclasts, which express the surface antigen F4/80 (142, 143), enter the circulation and arrive at the site to be resorbed. They will fuse together into a polykaryon (immature osteoclast) only in the presence of M-CSF and RANK-L. The immature osteoclast begins to express TRAP, calcitonin receptor, and the beta-3 integrin. RANK-L and a host of transcription factors are required to push the cell into a mature osteoclast phenotype, which maintains expression of many of the same immature osteoclast markers. ( b ) Osteoblasts are derived from a mesenchymal stem cell, which can also give rise to adipocytes, myoblasts, and chondrocytes. Proliferating precursors are pushed toward the preosteoblast phenotype by the expression of Runx2, Dlx5, and Msx2. The preosteoblast expresses collagen I and bone sialoprotein. Further, Runx2 expression, but also osterix, and members of the Wnt signaling cascade (b-catenin, TCF/LEF1) are required to achieve a mature, matrix-producing osteoblast phenotype (Col I, osteocalcin, and alkaline phosphatase expression). Osteoblasts that become trapped in the matrix express E11, an early osteocyte marker, and eventually express DMP-1, Mepe, and Sost as the mature osteocyte phenotype is reached. The red bars on the bottom of the figure indicate the expression sequence of several osteoblast/osteocyte gene promoter fragments that are used to drive expression of proteins at different stages of differentiation in the osteoblast lineage.
Physiological Functions and Application Areas of Osteoblast and Osteoclast Markers
Osteoblasts and osteoclasts are two types of cells that play crucial roles in the formation, maintenance, and remodeling of bone tissue. Osteoblasts are responsible for bone formation, while osteoclasts are involved in bone resorption. Here are the physiological functions and application areas of osteoblast and osteoclast markers:
Physiological Functions of Osteoblasts
- Bone Formation: Osteoblasts are responsible for synthesizing and depositing the organic matrix of bone, mainly composed of collagen type I. They also regulate mineralization by secreting proteins like osteocalcin, which binds calcium and promotes hydroxyapatite crystal formation.
- Bone Remodeling: Osteoblasts participate in bone remodeling by coordinating bone formation and resorption processes. They communicate with osteoclasts and other bone cells through signaling molecules, contributing to the balance between bone resorption and formation.
- Osteoid Production: Osteoblasts secrete osteoid, an unmineralized matrix rich in collagen and other proteins, which provides a scaffold for mineral deposition during bone formation.
- Regulation of Bone Cell Differentiation: Osteoblasts produce factors like osteoprotegerin (OPG) and RANKL (Receptor Activator of Nuclear Factor κB Ligand), which regulate the differentiation of osteoclasts from precursor cells and maintain the balance between bone formation and resorption.
Application Areas of Osteoblast Markers
- Bone Health and Diseases: Osteoblast markers are used to evaluate bone health and diagnose various bone diseases such as osteoporosis, osteogenesis imperfecta, and Paget's disease. Assessing markers like alkaline phosphatase (ALP) and osteocalcin (OCN) helps monitor bone formation and turnover rates.
- Evaluation of Bone Healing: Osteoblast markers are utilized to assess the progress and quality of bone healing in fracture repair and bone grafting procedures. Monitoring markers like ALP and collagen type I helps evaluate the activity of osteoblasts during the bone healing process.
- Bone Tissue Engineering: Osteoblast markers are employed to evaluate the functionality and maturation of engineered bone tissues. Assessing markers like ALP, OCN, and collagen type I helps determine the effectiveness of tissue engineering strategies and the integration of engineered constructs with the host bone.
Physiological Functions of Osteoclasts
- Bone Resorption: Osteoclasts are specialized cells responsible for bone resorption. They attach to bone surfaces and secrete enzymes, such as cathepsin K and matrix metalloproteinases (MMPs), which degrade the organic matrix and release calcium and other minerals.
- Bone Remodeling: Osteoclasts participate in the bone remodeling process by removing old or damaged bone tissue. They work in coordination with osteoblasts to maintain bone structure and strength.
Application Areas of Osteoclast Markers
- Bone Diseases: Osteoclast markers are used to evaluate bone diseases characterized by excessive bone resorption, such as osteoporosis, osteopetrosis, and Paget's disease. Monitoring markers like tartrate-resistant acid phosphatase (TRAP) and cathepsin K helps assess osteoclast activity and bone resorption levels.
- Therapeutic Interventions: Osteoclast markers are employed to evaluate the effectiveness of therapeutic interventions targeting bone resorption. Monitoring markers like TRAP and cathepsin K helps assess the impact of anti-resorptive drugs, such as bisphosphonates and denosumab, on osteoclast activity.
- Cancer Metastasis: Osteoclast markers are utilized in cancer research to study bone metastasis. Assessing osteoclast markers helps understand the role of osteoclasts in tumor-induced bone destruction and evaluate potential therapeutic strategies to inhibit osteoclast activity and bone resorption.
By utilizing osteoblast and osteoclast markers, researchers can gain insights into bone metabolism, bone remodeling, and the pathophysiology of bone diseases. These markers contribute to the diagnosis, monitoring, and development of therapeutic interventions for bone-related conditions, ultimately aiming to improve bone health and clinical outcomes.
Available Resources for Osteoblast and Osteoclast Markers
At Creative BioMart, we are dedicated to supporting scientific researchers in their study of osteoblast and osteoclast markers by providing a wide range of products and carefully tailored services. Our product line includes recombinant proteins and more, catering to various research needs. We have curated a vast collection of resources on osteoblast and osteoclast markers, covering topics such as involved pathways, protein functions, interaction proteins, relevant articles, research areas, and related subjects. Whether you are looking to gain a comprehensive understanding of the regulatory mechanisms and pathways of osteoblast and osteoclast markers or aiming to explore their roles and functions in different biological processes, we are well-equipped to meet your requirements.
Our Featured Products
- Recombinant Human OMD protein, His-tagged
- Recombinant Human IFNGR1 Protein, His-tagged
- Recombinant Human SPARC, GST-tagged
- Active Recombinant Human SPARC protein, His-tagged
- Recombinant Mouse Sparc, His tagged
- Recombinant Human TBX5, GST-tagged
- Recombinant Human REG3A, GST-tagged
- Recombinant Human REG3A, His tagged
- Recombinant Human BGN protein, His-tagged
- Recombinant Human CD44 protein, His-tagged
- Recombinant Human CD44, His-tagged
- Recombinant Human CD44 Protein, hFc-tagged, FITC conjugated
- Active Recombinant Mouse Cd44 Protein, Fc-tagged, Alexa Fluor 555 conjugated
- Active Recombinant Rat Cd44 Protein, Fc-tagged, FITC conjugated
- Active Recombinant Human ALCAM protein, His-tagged
- Active Recombinant Mouse Alcam protein, His-tagged
- Active Recombinant Mouse Alcam Protein, hIgG/His-tagged
- Active Recombinant Human TNFRSF11A protein, hFc-tagged
Should you have any questions, specific requirements, or intentions for collaboration, please don't hesitate to reach out to us. We eagerly anticipate the opportunity to collaborate with you and assist in achieving your research and commercial goals.
References:
- Robling A G, Castillo A B, Turner C H. Biomechanical and molecular regulation of bone remodeling[J]. Annual Review of Biomedical Engineering, 2006, 8(1):455-498.DOI:10.1021/ja903482u.
- Sheng Z, Sabrina E, Marc R, et al. From the Clinical Problem to the Basic Research — Co-Culture Models of Osteoblasts and Osteoclasts[J]. International Journal of Molecular Sciences, 2018, 19(8):2284-.DOI:10.3390/ijms19082284.