
Synaptic SNAREs
Related Symbol Search List
- SYT1
- CPLX1
- SNAPIN
- SNCA
- SYBU
- VAMP1
- EEA1
- SNAP23
- Ric-4
- SNAP29
- STX12
- STX16
- STX1A
- STX1B
- STX2
- STX4
- STX6
- STX7
- STX8
- STXBP1
- STXBP2
- STXBP3
- SV2C
- SYN1
- SYP
- TXLNA
- UNC13A
- UNC13B
- VAMP2
- VAMP5
- VAMP7
- VAMP8
- VAPA
- VAPB
Immunology Background
About Synaptic SNAREs
Synaptic SNAREs, also known as soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptors, are a group of proteins that play a crucial role in the process of neurotransmitter release at synapses. They are key components of the molecular machinery responsible for synaptic vesicle fusion and exocytosis.
Synaptic SNAREs are primarily located on the membranes of synaptic vesicles and the presynaptic plasma membrane of neurons. They function by forming a complex of intertwined protein helices known as the SNARE complex, which acts as a fusion clamp between the synaptic vesicle and plasma membrane. This complex consists of three core SNARE proteins, namely synaptobrevin (also called VAMP), syntaxin, and SNAP-25, which come from both the vesicle and presynaptic membrane.
The formation of the SNARE complex is a crucial step in synaptic vesicle fusion. It brings the vesicle and plasma membrane into close proximity, facilitating the merging of their lipid bilayers. This process ultimately leads to the release of neurotransmitters into the synaptic cleft, where they can then bind to receptors on the postsynaptic neuron and transmit the signal.
The specificity and efficiency of neurotransmitter release at synapses heavily rely on the precise regulation of SNARE complex formation. Various regulatory proteins and mechanisms work together to control the timing and extent of SNARE complex assembly, allowing for the precise control of synaptic vesicle fusion.
Overall, synaptic SNAREs play a fundamental role in synaptic transmission and the communication between neurons. Their intricate interactions and regulation ensure the proper functioning of the synapse and contribute to important physiological processes such as learning, memory formation, and neuronal development.
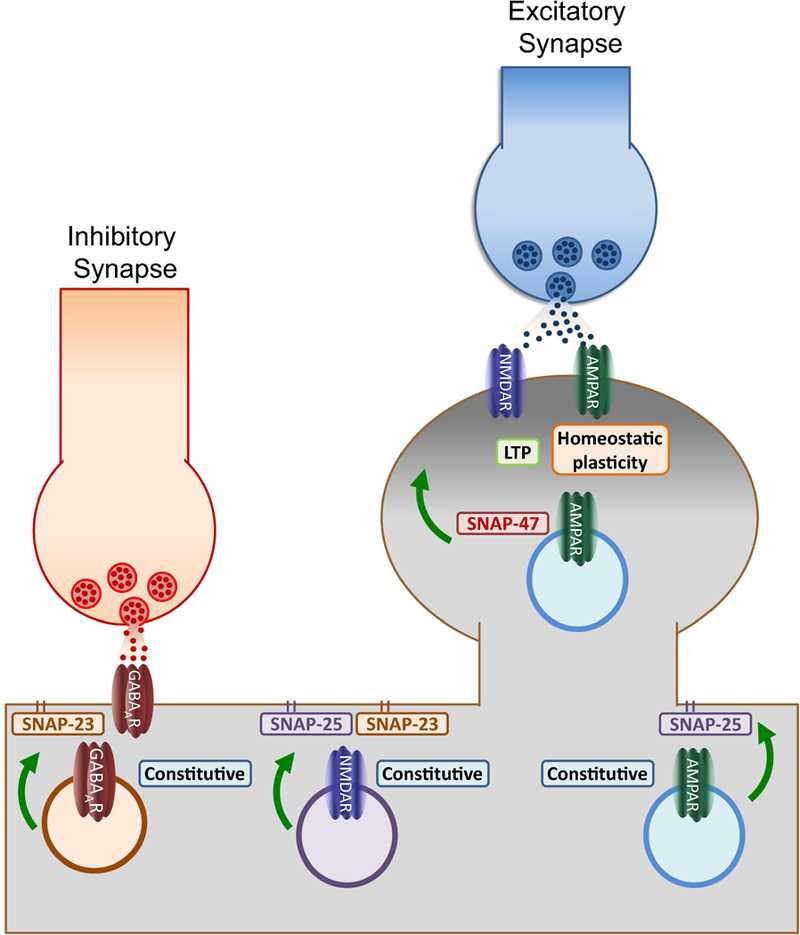 Fig.1 Functions of postsynaptic SNAPs. (Madrigal MP, 2018)
Fig.1 Functions of postsynaptic SNAPs. (Madrigal MP, 2018)
Schematic of the identified roles of postsynaptic SNAPs depicts membrane-bound SNAP-25 and SNAP-23 primarily involved in regulating constitutive trafficking of GABAA, AMPA and NMDA receptors. This function suggests the existence of specific membrane microdomains enriched with these SNAPs where exocytosis may be facilitated during baseline conditions. In contrast, activity-dependent insertion (NMDAR-dependent LTP and retinoic acid-induced potentiation) depends on the activity of SNAP-47, a broadly expressed cytosolic isoform. These results suggest that calcium influx increases dendritic exocytosis by recruiting SNAP isoforms like SNAP-47 which by not being attached to the membrane may define temporary regions for fusion along the dendritic membrane. By recruiting SNAP-47, the increase in postsynaptic exocytosis demand during synaptic plasticity can be supported in the absence of specialized regions like the presynaptic active zone.
Biological Functions of Synaptic SNAREs
Synaptic Vesicle Docking: Synaptic SNAREs, including syntaxin and SNAP-25 (synaptosomal-associated protein 25), participate in the initial docking of synaptic vesicles to the presynaptic membrane. Syntaxin, localized on the plasma membrane, forms a complex with SNAP-25 and other proteins, creating a docking site for the synaptic vesicle-associated SNARE protein, synaptobrevin/VAMP (vesicle-associated membrane protein). This interaction plays a crucial role in positioning synaptic vesicles close to the active zone for subsequent fusion.
Vesicle Priming: Synaptic SNAREs are involved in the process of vesicle priming, which prepares synaptic vesicles for fusion by making them competent to undergo exocytosis. During priming, the SNARE complex forms a partially assembled state, involving the interaction between syntaxin, SNAP-25, and synaptobrevin. This partially assembled SNARE complex provides the foundation for the subsequent rapid and efficient fusion of synaptic vesicles with the presynaptic membrane.
Fusion of Synaptic Vesicles: The primary function of synaptic SNAREs is to mediate the fusion of synaptic vesicles with the presynaptic membrane, leading to the release of neurotransmitters into the synaptic cleft. The fully assembled SNARE complex, consisting of syntaxin, SNAP-25, and synaptobrevin, brings the vesicle and plasma membrane in close proximity and drives the fusion process. This enables the release of neurotransmitters, which transmit signals across the synapse.
Regulation of Exocytosis: Synaptic SNAREs are involved in the regulation of exocytosis, ensuring the precise control of neurotransmitter release. The interactions between SNARE proteins and various regulatory proteins, such as Munc18 and Munc13, modulate the efficiency and kinetics of exocytosis. These regulatory proteins influence the assembly and disassembly of the SNARE complex, the availability of synaptic vesicles for fusion, and the calcium-dependent triggering of neurotransmitter release.
Synaptic Plasticity: Synaptic SNAREs play a role in synaptic plasticity, the ability of synapses to undergo activity-dependent changes in strength. The SNARE complex and its associated proteins can undergo modifications that regulate synaptic transmission and plasticity. For instance, phosphorylation of SNARE proteins, such as syntaxin and SNAP-25, by protein kinases can modulate the kinetics and efficacy of neurotransmitter release, contributing to the synaptic plasticity underlying learning and memory processes.
Neurological Disorders: Dysfunction or dysregulation of synaptic SNAREs has been implicated in various neurological disorders. Mutations or alterations in SNARE proteins, as well as the proteins involved in their regulation, can disrupt synaptic vesicle fusion and neurotransmitter release, leading to synaptic dysfunction. Abnormal synaptic SNARE function has been associated with conditions such as epilepsy, Parkinson's disease, and neurodevelopmental disorders, highlighting the importance of these proteins in maintaining normal synaptic function.
In summary, synaptic SNAREs have critical biological functions in synaptic vesicle docking, priming, fusion, and the regulation of neurotransmitter release. They play a pivotal role in synaptic transmission and are involved in synaptic plasticity and various neurological disorders. Understanding the functions and regulation of synaptic SNAREs provides insights into the mechanisms of synaptic communication and has implications for the development of therapeutic strategies targeting synaptic dysfunction.
The Application Areas of Synaptic SNAREs
Neurotransmitter Release Studies: Synaptic SNAREs are widely used in research aimed at understanding the mechanisms of neurotransmitter release. By manipulating the expression or function of specific SNARE proteins, researchers can investigate their roles in synaptic vesicle fusion and neurotransmission. These studies provide insights into the fundamental processes underlying synaptic function and synaptic transmission.
Neurological Disorder Research: Dysfunction of synaptic SNAREs has been implicated in several neurological disorders, including epilepsy, Parkinson's disease, and neurodevelopmental disorders. Research focuses on studying the alterations in SNARE protein expression, localization, or activity in these disorders to better understand their contributions to disease pathogenesis. Investigating synaptic SNAREs in neurological disorders may offer potential diagnostic markers, therapeutic targets, or avenues for developing novel treatment strategies.
Drug Development: Synaptic SNAREs and their associated proteins represent potential targets for drug development. Modulating the function or interactions of SNARE proteins can influence synaptic vesicle fusion and neurotransmitter release, making them attractive targets for developing therapeutic interventions. Small molecules, peptides, or antibodies that selectively interact with synaptic SNAREs or modulate their regulatory proteins are being explored for their potential in treating neurological disorders or improving synaptic function.
Neuroimaging: Synaptic SNAREs are utilized in neuroimaging research to investigate synaptic integrity and function in vivo. Radioligands or contrast agents that specifically target synaptic SNARE proteins can be used in imaging techniques such as positron emission tomography (PET) or magnetic resonance imaging (MRI). These imaging approaches provide insights into synaptic abnormalities, synaptic density, and changes in synaptic function associated with neurological disorders.
Biomarkers for Neurological Disorders: Alterations in synaptic SNAREs and associated proteins may serve as biomarkers for neurological disorders. Changes in the levels or activity of synaptic SNAREs in patient samples, such as cerebrospinal fluid or blood, can provide indications of synaptic dysfunction or disease progression. Measuring synaptic SNARE biomarkers may aid in the diagnosis, monitoring, or prognostic assessment of neurological disorders.
Synaptic Plasticity and Learning: Synaptic SNAREs are involved in synaptic plasticity, which underlies learning and memory processes. Research on synaptic SNAREs and related proteins contributes to our understanding of the molecular mechanisms that regulate synaptic plasticity. Manipulating synaptic SNARE function or interactions may offer opportunities for enhancing synaptic plasticity and improving learning and memory.
Neuroengineering and Neural Interfaces: Synaptic SNAREs are relevant to the field of neuroengineering and the development of neural interfaces. Incorporating synaptic SNAREs into bioelectrodes or biomaterials can facilitate the integration and communication between artificial devices and neural tissue. Understanding the interactions and dynamics of synaptic SNAREs can contribute to the design and development of more effective brain-machine interfaces or neural prosthetic devices.
The application areas of synaptic SNAREs encompass a wide range of research and clinical domains, including neurotransmitter release studies, neurological disorder research, drug development, neuroimaging, biomarker discovery, synaptic plasticity, and neuroengineering. Continued investigation of synaptic SNAREs offers valuable insights into synaptic function, synaptic dysfunction in neurological disorders, and potential avenues for therapeutic interventions.
Available Resources for Synaptic SNAREs
Creative BioMart is committed to providing scientists with high-quality research tools that enable them to make breakthroughs in the field of synaptic protein research. Our product line extensively covers recombinant proteins related to Synaptic SNAREs, cell and tissue lysates, and protein pre-coupled magnetic beads. In addition, we provide customized services based on customer needs, including protein expression, purification and custom development. and provides a wide range of educational and technical resources designed to help researchers better understand and apply synaptic protein research.
Feel free to view the synaptic protein-related molecules below and click for more comprehensive resources.
Whether you are interested in a specific product or have additional questions, our team is here to support you. If you require detailed information, custom services, or more related resources, please feel free to contact us. We look forward to collaborating with you on research in the field of Synaptic SNAREs and jointly promoting scientific progress!
References:
- Madrigal MP, Portalés A, SanJuan MP, Jurado S. Postsynaptic SNARE Proteins: Role in Synaptic Transmission and Plasticity. Neuroscience. 2019;420:12-21. doi:10.1016/j.neuroscience.2018.11.012.
- Cali E, Rocca C, Salpietro V, Houlden H. Epileptic Phenotypes Associated With SNAREs and Related Synaptic Vesicle Exocytosis Machinery. Front Neurol. 2022;12:806506. Published 2022 Jan 13. doi:10.3389/fneur.2021.806506