
What is CD163 Protein
The CD163 protein, also referred to as Cluster of Differentiation 163, was initially discovered in 1987. A member of the scavenger receptor cysteine-rich (SRCR) superfamily, the CD163 protein is encoded by the CD163 gene located on the gene locus 12p13.3. Significant for its role in immune regulation and inflammation, CD163 boasts an extensive structure, functioning in multiple biological pathways, and has transformative applications in biomedical science.
Although it was first recognized in 1987 from activated monocytes, an in-depth understanding of the CD163 protein and its functional significance only became apparent in the early 2000s. With subsequent genetic analysis, its gene locus was determined as 12p13.3. The protein is predominantly expressed in the monocyte-macrophage system, a part of the body's immune response.
CD163 Protein Structure
The structural complexity of the CD163 protein is instrumental in performing its various functions. It is characterized by a large extracellular domain with nine SRCR, a single transmembrane domain, and a small cytoplasmic tail. The SRCR domains selectively bind haptoglobin-hemoglobin (Hp-Hb) complexes due to their highly-ligand-binding capacity. The protein uses this binding ability to perform its function, thereby contributing to its immune regulatory role.
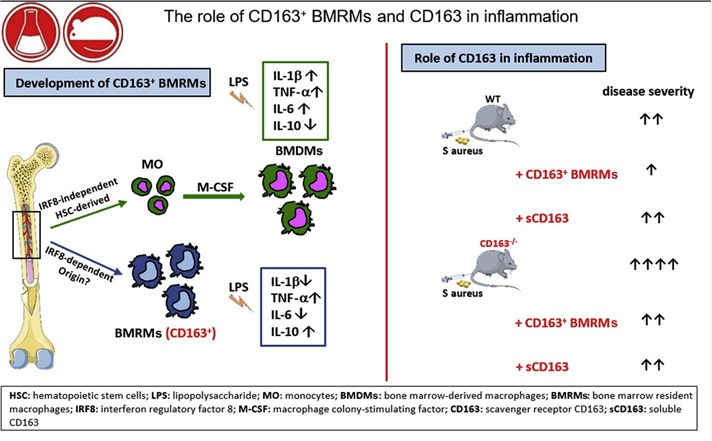
Fig1. The role of CD163 in inflammation (Fischer-Riepe, L., et al, 2020)
Function of CD163 Protein
The CD163 protein mainly functions as an anti-inflammatory marker and is responsible for regulating our body's immune responses. The protein achieves this through the main scavenger function, by which it binds and ingests hemoglobin (Hb) released during hemolysis. Once Hb is bound to haptoglobin (Hp), the Hp-Hb complex is recognized and ingested by the CD163 protein, thereby protecting tissues from harmful Hb exposure.
CD163 Protein-related Signal Pathway
In the signal transduction pathway, the CD163 protein has been implicated in several anti-inflammatory pathways. CD163 upregulation leads to the activation of signaling pathways involved in the anti-inflammatory cytokine interleukin-10 (IL-10). It also inhibits tumor necrosis factor-alpha (TNF-α) production. Additionally, CD163 cleaves the Hp-Hb complex, releasing an anti-inflammatory molecule known as heme, which subsequently stimulates heme oxygenase-1 (HO-1). This contributes to tissue protection and anti-inflammatory response.
CD163 Protein-Related Diseases
Given its central immune regulatory role, the dysregulation of CD163 protein is associated with several diseases. For instance, elevated levels of the protein have been observed in several pathologies characterized by inflammation, including sepsis, autoimmune diseases, and cancer. In cancer, specifically, CD163 overexpression confers a poor prognosis, with tumors often being more aggressive. Moreover, research has linked high CD163 levels to HIV infection and its progression to AIDS, emphasizing the protein's importance in both immune and infectious diseases.
CD163 Protein in Biomedicine
The CD163 protein has profound potential in biomedicine. Its overexpression in several pathological conditions makes it an excellent indicator for diagnostic and therapeutic applications. As a biomarker, CD163 can aid in diagnosing and monitoring inflammatory conditions, infectious diseases, and cancer. In immunotherapy, antagonists to CD163 are under exploration to modulate the immune response. Specific antibodies can help identify and target cells overexpressing CD163, thus having great therapeutic potential in treating diseases such as cancer and HIV.
The CD163 protein, with its complex structure and crucial role in immune regulation, significantly impacts biological pathways and disease states. While we have made meaningful progress in our understanding of this critical protein, much remains to be discovered. Its association with various diseases, coupled with its potential biomedical applications, underscores the importance of continued research in this field as we move towards developing more targeted therapeutic approaches.
Our Featured Products
| Cat.No. | Product Name | Species | Source (Host) | Tag |
|---|---|---|---|---|
| CD163-112H | Active Recombinant Human CD163, His-tagged | Human | Mammalian cells | His |
| CD163-27220TH | Recombinant Human CD163 | Human | Wheat Germ | N/A |
| CD163-10910H | Recombinant Human CD163, GST-tagged | Human | E.coli | GST |
| CD163-356H | Recombinant Human CD163 protein, His-tagged | Human | HEK293 | His |
| CD163-2625H | Recombinant Human CD163 Protein, His (Fc)-Avi-tagged | Human | HEK293 | His (Fc)-Avi |
| CD163-1463HFL | Recombinant Full Length Human CD163 Protein, C-Flag-tagged | Human | Mammalian cells | Flag |
| Cd163-1436M | Recombinant Mouse Cd163 Protein, His (Fc)-Avi-tagged | Mouse | HEK293 | His (Fc)-Avi |
| Cd163-371M | Active Recombinant Mouse Cd163, His-tagged | Mouse | Mammalian cells | His |
Reference
- Fischer-Riepe, L., Daber, N., Schulte-Schrepping, J., Véras De Carvalho, B. C., Russo, A., Pohlen, M., Fischer, J., Chasan, A. I., Wolf, M., Ulas, T., Glander, S., Schulz, C., Skryabin, B., Wollbrink, Dipl-Ing, A., Steingraeber, N., Stremmel, C., Koehle, M., Gärtner, F., Vettorazzi, S., . . . Barczyk-Kahlert, K. (2020). CD163 expression defines specific, IRF8-dependent, immune-modulatory macrophages in the bone marrow. Journal of Allergy and Clinical Immunology, 146(5), 1137-1151. https://doi.org/10.1016/j.jaci.2020.02.034