



MAPS-A Novel Computational Tool for Enhanced Analysis of Chromatin Interactions
Chromatin spatial organization plays a pivotal role in gene regulation, with technologies like PLAC-seq and HiChIP enabling efficient detection of long-range interactions anchored at protein-binding sites. However, existing computational tools for analyzing these datasets often fail to account for biases introduced during chromatin immunoprecipitation (ChIP), leading to suboptimal sensitivity and accuracy. The article introduces MAPS (Model-based Analysis of PLAC-seq and HiChIP), a computational pipeline designed to overcome these limitations and improve the identification of biologically relevant chromatin interactions.
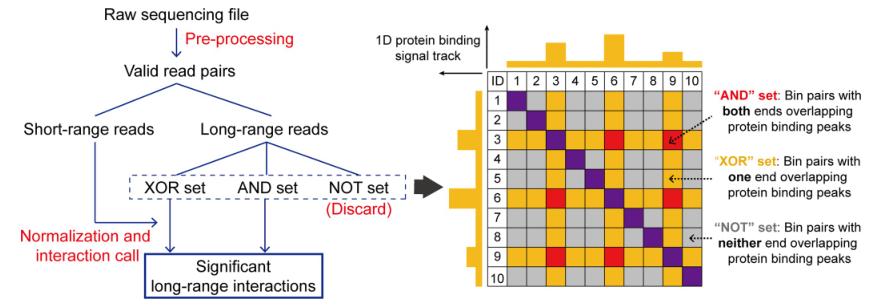
MAPS addresses two major challenges in PLAC-seq/HiChIP data analysis:
- ChIP-Specific Biases: Unlike Hi-C normalization methods (e.g., ICE or KR), which assume equal visibility of genomic regions, MAPS incorporates ChIP enrichment levels into a zero-truncated Poisson regression model. This corrects for biases such as variable protein-binding efficiency and fragment length.
- Detection of Diverse Interactions: Existing tools like Mango and hichipper focus only on interactions where both ends are protein-bound ("AND" set). MAPS additionally identifies interactions where only one end is bound ("XOR" set), significantly expanding the scope of detectable regulatory connections.
The MAPS workflow involves:
- Pre-processing: Filtering raw sequencing data, classifying interactions into "AND," "XOR," or "NOT" sets.
- Normalization: Separately modeling "AND" and "XOR" interactions to account for differing contact frequencies.
- Interaction Calling: Applying statistical thresholds (e.g., FDR < 0.01, normalized contact frequency > 2) to identify significant interactions.
When tested on four datasets (GM12878 HiChIP and mESC PLAC-seq), MAPS demonstrated superior performance:
- Higher Sensitivity: MAPS recovered 91.8–97.6% of HiCCUPS-validated loops from deeply sequenced Hi-C data, outperforming hichipper (32.8–68.5%).
- Biological Relevance: MAPS-identified interactions showed stronger enrichment for CTCF convergent motifs (up to 76.7% vs. hichipper's 67%) and overlapped with cis-regulatory elements (enhancers, promoters) and highly expressed genes.
- Novel Discoveries: MAPS detected interactions missed by HiCCUPS, validated by orthogonal methods like SPRITE and CRISPR-based enhancer studies.
By integrating ChIP-specific normalization and expanding the scope of detectable interactions, MAPS addresses critical gaps in chromatin interaction analysis. Its ability to uncover previously overlooked regulatory connections—validated through multiple lines of evidence—positions it as a vital tool for advancing epigenomics and 3D genome research. Future developments, such as inter-chromosomal interaction analysis and allelic-specific profiling, could further enhance its utility in unraveling the complexities of genome organization.
Reference
- Juric I, Yu M, Abnousi A, Raviram R, Fang R, Zhao Y, Zhang Y, Qiu Y, Yang Y, Li Y, Ren B, Hu M. MAPS: Model-based analysis of long-range chromatin interactions from PLAC-seq and HiChIP experiments. PLoS Comput Biol. 2019 Apr 15;15(4):e1006982. doi: 10.1371/journal.pcbi.1006982. PMID: 30986246; PMCID: PMC6483256.
Contact us or send an email at for project quotations and more detailed information.
Quick Links
-

Papers’ PMID to Obtain Coupon
Submit Now -

Refer Friends & New Lab Start-up Promotions