
IL-12 Signaling Related Molecules
Related Symbol Search List
Immunology Background
Available Resources for IL-12 Signaling Related Molecules Research
Creative BioMart is delighted to showcase our diverse collection of products associated with IL-12 signaling-related molecules. Our extensive product portfolio includes high-quality recombinant proteins, protein pre-coupled magnetic beads, cell and tissue lysates, and more. These indispensable tools cater to the diverse needs of researchers, equipping them to conduct comprehensive studies in their respective fields. With customizable services, we ensure that you find the perfect product tailored to your specific research requirements.
In addition to our comprehensive product offering, we are dedicated to providing a wealth of information on IL-12 signaling-related molecules. Our resources cover a broad spectrum of topics, including pathways, protein function, interacting proteins, related articles, and research areas. These valuable resources serve as references for researchers seeking to enhance their understanding of IL-12 signaling-related molecules and their pivotal roles in various physiological processes. By offering an integration of products and knowledge, we actively support researchers in driving advancements within this dynamic research field.
Our Featured Products
| Cat.# | Product name | Species | Source (Host) | Tag |
|---|---|---|---|---|
| STAT1-7037H | Recombinant Human STAT1 protein (Met1-Val712), His&GST-tagged | Human | Insect Cells | N-His&GST |
| STAT1-2996H | Recombinant Human STAT1 protein, GST-tagged | Human | E.coli | GST |
| STAT3-1496H | Recombinant Human STAT3, GST-tagged | Human | Sf9 Insect Cell | GST |
| STAT3-29823TH | Recombinant Human STAT3, His-tagged | Human | E.coli | His |
| STAT3-39228H | Recombinant Human STAT3, GST-tagged | Human | E.coli | GST |
About IL-12 Signaling Related Molecules
The IL-12 family of cytokines consists of IL-12, IL-23, and IL-27, which play crucial roles in regulating immune responses and inflammation. The signaling pathway of the IL-12 family involves several key molecules, including STAT1, STAT3, and STAT4. Let's explore these molecules and their roles in IL-12 family signaling:
Signal Transducer and Activator of Transcription 1 (STAT1):
STAT1 is a transcription factor that plays a central role in the signaling pathways of IL-12, IL-23, and IL-27. Upon stimulation by IL-12 or IL-27, JAK2, and Tyk2 kinases are activated and phosphorylate specific tyrosine residues on the cytoplasmic tail of the receptor subunits. This leads to the recruitment and phosphorylation of STAT1 by JAK kinases. Phosphorylated STAT1 proteins form homodimers or heterodimers with other STAT proteins and translocate to the nucleus. Within the nucleus, STAT1 dimers bind to specific DNA sequences known as gamma-activated sites (GAS) in the promoter regions of target genes, resulting in the transcriptional regulation of genes involved in immune responses and inflammation.
Signal Transducer and Activator of Transcription 3 (STAT3)
STAT3 is another important transcription factor involved in IL-6 family cytokine signaling, including IL-23 signaling. Upon IL-23 stimulation, JAK2 and Tyk2 kinases are activated and phosphorylate specific tyrosine residues on the receptor subunits. This leads to the recruitment and phosphorylation of STAT3 by JAK kinases. Phosphorylated STAT3 proteins form homodimers or heterodimers and translocate to the nucleus, where they regulate the transcription of target genes involved in immune responses, inflammation, and cell survival.
Signal Transducer and Activator of Transcription 4 (STAT4)
STAT4 is specifically activated by IL-12 signaling. Following IL-12 binding to its receptor, JAK2 and Tyk2 kinases phosphorylate the cytoplasmic tail of the receptor subunits, leading to the recruitment and activation of STAT4. Phosphorylated STAT4 proteins form homodimers and translocate to the nucleus, where they bind to specific DNA sequences known as IL-12 response elements (IL-12RE) in the promoter regions of target genes. STAT4 activation is crucial for the differentiation of Th1 cells, which play a key role in cellular immune responses, production of IFN-γ, and defense against intracellular pathogens.
Understanding the roles of STAT1, STAT3, and STAT4 in the signaling pathways of the IL-12 family of cytokines provides insights into the regulation of immune responses, inflammation, and the development of specific T-cell subsets.
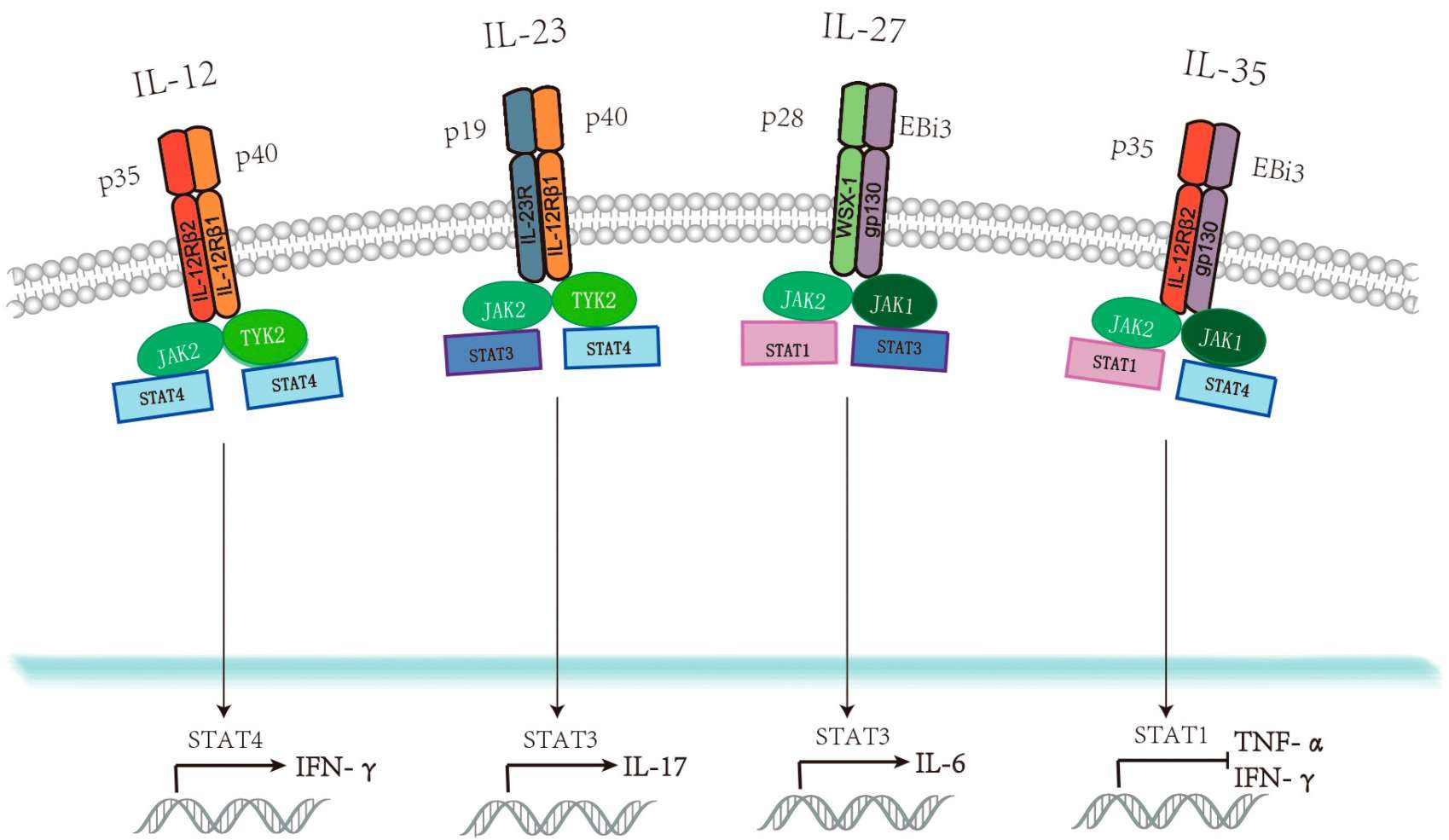 Fig.1 Interleukin 12 (IL-12) family members, corresponding receptors, and regulation of downstream signaling pathways. (Guo Y, et al., 2019)
Fig.1 Interleukin 12 (IL-12) family members, corresponding receptors, and regulation of downstream signaling pathways. (Guo Y, et al., 2019)
Role of IL-12 Signaling Related Molecules in Disease Development
The IL-12 family signaling-related molecules, including STAT1, STAT3, and STAT4, play crucial roles in immune responses and inflammation, and their dysregulation can contribute to the development of various diseases. Here are some examples of how these molecules are involved in disease development:
STAT1
- Autoimmune Diseases: Dysregulated STAT1 signaling has been associated with autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA). Increased STAT1 activation can lead to the production of pro-inflammatory cytokines and promote autoimmunity.
- Infectious Diseases: STAT1 deficiency or impaired STAT1 signaling can result in susceptibility to intracellular bacterial or viral infections. Proper STAT1 activation is necessary for the induction of antimicrobial responses and the production of IFN-γ, which are important for controlling infections.
STAT3
- Inflammatory Bowel Diseases (IBD): Dysregulated STAT3 signaling has been implicated in the development of IBD, including Crohn's disease and ulcerative colitis. Excessive activation of STAT3 can lead to increased production of pro-inflammatory cytokines and contribute to chronic inflammation in the gut.
- Cancer: Aberrant STAT3 activation is frequently observed in various cancers. Persistent STAT3 signaling promotes tumor cell survival, proliferation, angiogenesis, and immune evasion. Targeting STAT3 signaling has emerged as a potential therapeutic strategy for cancer treatment.
STAT4
- Autoimmune Diseases: Dysregulated STAT4 signaling has been associated with autoimmune diseases, including multiple sclerosis (MS) and systemic sclerosis (SSc). Increased STAT4 activation can contribute to the dysregulation of immune responses and the promotion of autoimmunity.
- Infectious Diseases: Proper STAT4 activation is essential for effective immune responses against intracellular pathogens. Impaired STAT4 signaling can result in increased susceptibility to infections, such as tuberculosis and viral infections.
It's important to note that the roles of these molecules in disease development can vary depending on the specific context and disease condition. Additionally, other factors and signaling pathways also contribute to the pathogenesis of diseases. Further research is needed to fully understand the complex interactions and mechanisms underlying the involvement of IL-12 family signaling-related molecules in disease development.
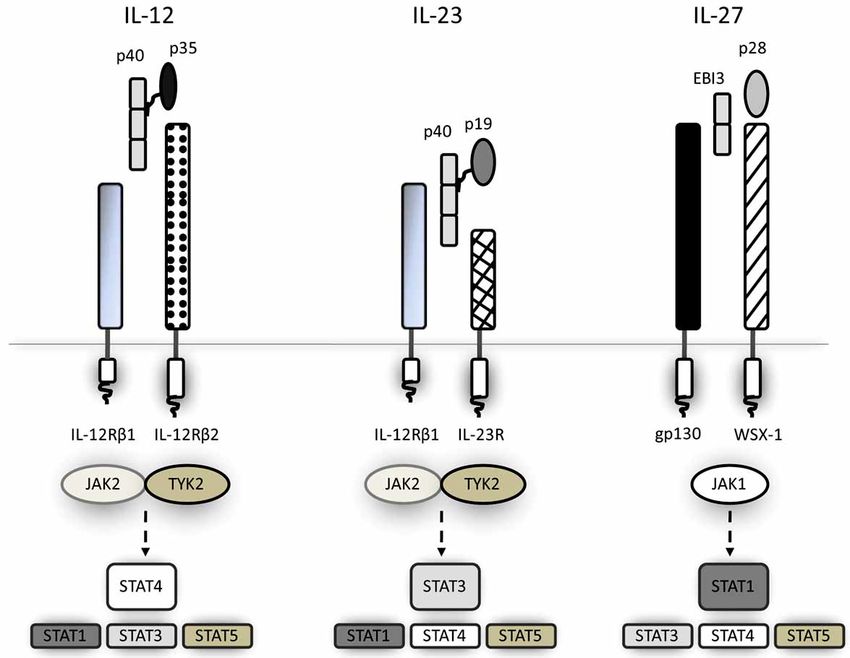 Fig.2 Receptors and JAK/STAT signalling pathways for the IL-12 family cytokines. (Gee K, et al., 2009)
Fig.2 Receptors and JAK/STAT signalling pathways for the IL-12 family cytokines. (Gee K, et al., 2009)
The IL-12 receptor which has homology with gp130, consists of IL-12R1 and IL-12R2. IL-12 is a heterodimeric cytokine, composed of a light chain (IL-12p35) and a heavy chain (IL-12p40) which are covalently linked together. The association of IL-12R1 and IL-23R also form a receptor for another heterodimeric cytokine, IL- 23. The IL-12p40 component of IL-12 can also dimerize with IL-23p19 to form IL-23. IL-27, composed of EBI3 and IL-27p28, binds a receptor composed of gp130 and WSX-1. The main signalling pathway induced by the IL-12 family cytokines is the JAK/STAT pathway and the specific molecules activated by the each cytokine are shown. As indicated by the enlarged text, IL-12 mainly activates STAT4, whereas IL-23 and IL-27 mainly activate STAT3 and STAT1, respectively.
If you have any questions, requirements, or cooperation intentions, please feel free to contact us. We very much look forward to working with you and helping you achieve research and commercial success.
Related References:
- Guo Y, Cao W, Zhu Y. Immunoregulatory Functions of the IL-12 Family of Cytokines in Antiviral Systems. Viruses. 2019; 11(9):772. https://doi.org/10.3390/v11090772
- Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm Allergy Drug Targets. 2009;8(1):40-52. doi:10.2174/187152809787582507