



SC-PSILAC, Single-Cell Proteomics using Stable Isotope Labeling by Amino acids in Cell Culture
Protein turnover-the balance of synthesis and degradation—is fundamental to cellular adaptation, yet its study has long been constrained by bulk analyses that mask single-cell variability. SC-pSILAC, a groundbreaking approach that integrates pulsed isotopic labeling with advanced single-cell proteomics. This method not only quantifies protein abundance but also tracks turnover rates, offering a multidimensional lens into cellular physiology.
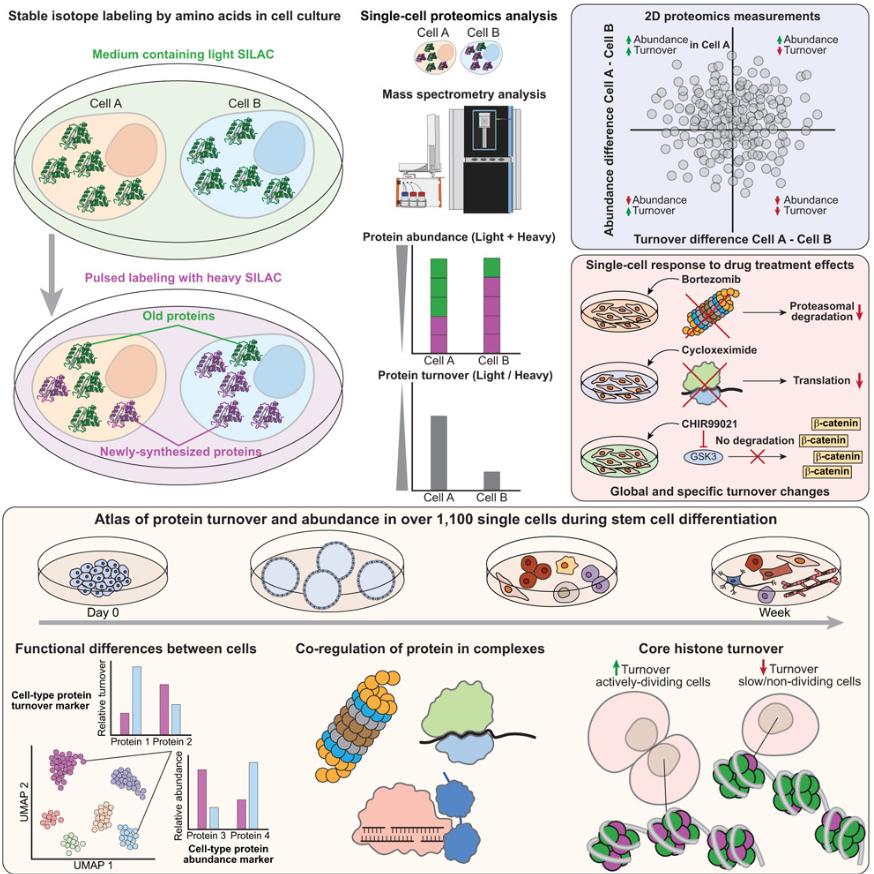
The SC-pSILAC method leverages heavy isotope-labeled amino acids to distinguish newly synthesized proteins from existing ones. By analyzing the light/heavy isotope ratio, the team demonstrated robust correlations between single-cell and bulk data in HeLa and HEK293T cells. Drug perturbations further validated the method: bortezomib, a proteasome inhibitor, increased histone stability, while cycloheximide suppressed translation, both detectable at the single-cell level. These findings underscore SC-pSILAC's precision in capturing pharmacodynamic responses and cellular heterogeneity.
Applying SC-pSILAC to iPSC differentiation revealed dynamic protein turnover patterns. Dividing cells exhibited rapid histone turnover, while non-dividing cells showed stable histones, correlating with chromatin compaction. This distinction, invisible in bulk analyses, highlights SC-pSILAC's ability to resolve subpopulations—critical for understanding stem cell fate transitions. Notably, cell-cycle proteins like MCM3 were more abundant in smaller, actively dividing cells, challenging the notion that protein abundance scales uniformly with cell size.
The study emphasizes that turnover rates provide complementary insights to abundance measurements. For instance, antioxidant enzymes in hepatocytes exhibited slower turnover (indicating stability) without abundance changes, a finding masked by transcriptomics. Similarly, co-regulation of proteasome subunits and histones revealed complex-specific dynamics, illustrating how turnover data can elucidate protein complex assembly and function.
SC-pSILAC's ability to distinguish quiescent (contact-inhibited) from stressed (serum-deprived) cells has profound implications for cancer research, where dormant tumor cells evade therapies. The method's resolution of histone dynamics could aid in identifying therapeutic targets in senescence or regenerative medicine. However, limitations remain, including challenges in clinical translation (due to isotopic labeling requirements) and the need for absolute synthesis/degradation rate measurements.
By linking protein stability to functional states-such as division, differentiation, and stress-SC-pSILAC opens avenues for precision medicine and mechanistic studies. Future advancements, such as multiplexed labeling and improved computational workflows, could further enhance its scalability, cementing its role as a cornerstone in systems biology.
Reference
- Sabatier P, Lechner M, Guzmán UH, Beusch CM, Zeng X, Wang L, Izaguirre F, Seth A, Gritsenko O, Rodin S, Grinnemo KH, Ye Z, Olsen JV. Global analysis of protein turnover dynamics in single cells. Cell. 2025 Mar 26:S0092-8674(25)00275-2. doi: 10.1016/j.cell.2025.03.002. Epub ahead of print. PMID: 40168994.
Contact us or send an email at for project quotations and more detailed information.
Quick Links
-

Papers’ PMID to Obtain Coupon
Submit Now -

Refer Friends & New Lab Start-up Promotions