



ChIP-DIP technology opens up a new era of protein DNA interactions
ChIP-DIP (Chromatin Immunoprecipitation Done in Parallel), a groundbreaking method developed by researchers at the California Institute of Technology and collaborators to simultaneously map hundreds of DNA-associated proteins in a single experiment. Published in Nature Genetics, this technology addresses the limitations of traditional protein-DNA mapping techniques, which are restricted to analyzing one protein at a time, and enables large-scale, cell-type-specific profiling of chromatin regulators, transcription factors (TFs), and histone modifications.
ChIP-DIP combines antibody-bead complexes tagged with unique oligonucleotides, split-pool barcoding, and high-throughput sequencing. This allows parallel immunoprecipitation of chromatin bound to hundreds of proteins, followed by computational deconvolution to generate individual protein-DNA interaction maps. The method is robust across diverse protein classes (histone modifications, chromatin regulators, TFs) and cell types, requiring as few as 50,000 cells while maintaining accuracy comparable to traditional ChIP-seq.
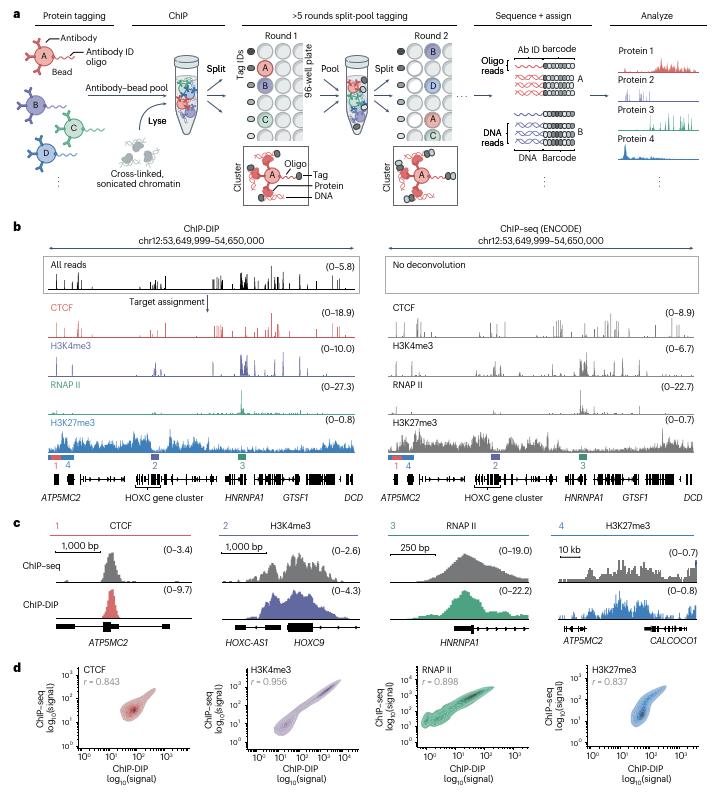
ChIP-DIP profiles matched ENCODE consortium data for proteins like CTCF, H3K4me3, and RNA polymerase II, with high genome-wide correlations (r=0.84–0.96r=0.84–0.96). Scalability was demonstrated by mapping >160 proteins in mouse embryonic stem cells (mESCs) and human K562 cells, including low-abundance TFs and RNA polymerase isoforms.
In LPS-stimulated dendritic cells, ChIP-DIP revealed temporal changes in histone modifications (e.g., H3K27ac at inflammatory genes) and linked enhancer dynamics to transcriptional activation. Combinatorial histone modification patterns defined distinct promoter and enhancer states, such as "bivalent domains" (H3K4me3 + H3K27me3) and stress-responsive enhancers marked by H2A.Z acetylation.
Biological Insights
- Promoter Diversity: RNA polymerase (I, II, III) promoters exhibited unique histone modification landscapes (e.g., H3K56ac enrichment at RNAP I/III genes).
- Enhancer Classification: Non-negative matrix factorization identified five enhancer classes, including pluripotency-associated (C4) and differentiation-poised (C5) elements, each with distinct TF and chromatin regulator binding.
- Regulatory Plasticity: Enhancer acetylation patterns correlated with gene expression changes, highlighting enhancers—not promoters—as primary drivers of transcriptional responses.
Advantages Over Existing Methods
- Scalability: Maps hundreds of proteins in one experiment, overcoming the throughput limitations of ChIP-seq and multiplexed CUT&Tag.
- Versatility: Applicable to diverse proteins (e.g., low-abundance TFs) and cell types, including primary cells.
- Cost-Efficiency: Reduces cell input and labor by orders of magnitude compared to traditional approaches.
ChIP-DIP democratizes large-scale epigenomic profiling, enabling individual labs to generate comprehensive regulatory maps without consortium-scale resources. It opens avenues to study dynamic gene regulation in disease models, rare cell populations, and developmental systems. Future integrations with 3D genomics or single-cell technologies could further illuminate the interplay between protein binding, chromatin architecture, and transcription.
By bridging the gap between single-protein assays and systems-level epigenomics, ChIP-DIP redefines our capacity to decode the regulatory genome. Its modular design and scalability position it as a transformative tool for unraveling the complexity of gene regulation in health and disease.
Reference
- Perez, A.A., Goronzy, I.N., Blanco, M.R. et al. ChIP-DIP maps binding of hundreds of proteins to DNA simultaneously and identifies diverse gene regulatory elements. Nat Genet 56, 2827–2841 (2024). https://doi.org/10.1038/s41588-024-02000-5
Contact us or send an email at for project quotations and more detailed information.
Quick Links
-

Papers’ PMID to Obtain Coupon
Submit Now -

Refer Friends & New Lab Start-up Promotions