
The p53 Pathway
🧪 KRAS-2569H
Source: E.coli
Species: Human
Tag:
Conjugation:
Protein Length: 1-185 aa
🧪 AKT3-131H
Source: Insect Cells
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-479 a.a.
🧪 CDK2-615H
Source: Insect Cells
Species: Human
Tag:
Conjugation:
Protein Length: Met 1-Leu 298
🧪 AKT1-786H
Source: Insect Cells
Species: Human
Tag: His
Conjugation:
Protein Length: 1-480 aa
🧪 CCNE1-3075H
Source: Insect Cells
Species: Human
Tag:
Conjugation:
Protein Length: Met1-Ala410
🧪 CDK1-3330H
Source: Insect Cells
Species: Human
Tag:
Conjugation:
Protein Length: Met 1-Met 297
🧪 CDK4-758H
Source: Insect Cells
Species: Human
Tag:
Conjugation:
Protein Length: Met1-Glu303
🧪 GADD45A-3880H
Source: Insect Cells
Species: Human
Tag:
Conjugation:
Protein Length: Met1-Arg165
🧪 PTEN-1619H
Source: E.coli
Species: Human
Tag:
Conjugation:
Protein Length: 1-403 a.a.
$99.00
$198
/ 5μg$149.00
$298
/ 10μg.jpg)
🧪 ARF1-9799H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: Met1-Lys181
🧪 ARF3-9800H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-181a.a.
🧪 ARF4-9801H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-180a.a.
🧪 ARF5-9802H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1-180a.a.
🧪 ARF6-9803H
Source: E.coli
Species: Human
Tag: His
Conjugation:
Protein Length: 37-152a.a.
🧪 ATM-9987H
Source: E.coli
Species: Human
Tag: GST
Conjugation:
Protein Length: 1332-1479a.a.
Background of The p53 Pathway
The p53 signaling pathway is a complex biological signaling network, which plays a key role in cell response to DNA damage, cell cycle control, apoptosis, cell metabolism, and maintenance of genome stability. p53, a tumor suppressor protein known as the "guardian of the genome," is known for its importance in preventing cancer from developing.
Mechanistically, a stress signal is transmitted to the p53 protein by post-translational modifications, which activates the p53 protein as a transcription factor that initiates a program of cell cycle arrest, cellular senescence, or apoptosis. The transcriptional network of p53-responsive genes produces proteins that interact with many other signal transduction pathways in the cell, and a number of positive and negative auto-regulatory feedback loops act upon the p53 response. To further elaborate, the p53 circuit communicates with the Wnt-beta-catenin, IGF-1-AKT, Rb-E2F, p38 MAP kinase, cyclin-CDK, p14/19 ARF pathways, and the cyclin G-PP2A and p73 gene products.
The two main apoptosis pathways of p53 signaling pathway include extrinsic pathway and intrinsic pathway. The extrinsic pathway involves the activation of "death" receptors belonging to the tumor necrosis factor receptor (TNFR) family that activate caspase enzymes, such as Caspase8 and Caspase3, by forming a death inducing signaling complex (DISC) that induces apoptosis. The intrinsic pathway involves the regulation of Bcl-2 family proteins, which control the permeability of the mitochondrial outer membrane and thus affect the apoptosis process.
What is p53?
p53 is an important tumor suppressor gene. Since the discovery of the p53 gene in 1979, over 80,000 research papers have been reported. Initially, the p53 gene was thought to be an oncogene, but with the deepening of research during the past decades, the function of p53 as a tumor suppressor was gradually revealed. Human p53 is active as a homotetramer of 4 × 393 amino acids with a complex domain organization, consisting of an intrinsically disordered N-terminal transactivation domain (TAD), a proline (Pro)-rich region, a structured DNA-binding domain (DBD), and tetramerization domain connected via a flexible linker, and an intrinsically disordered C-terminal regulatory domain. The p53 tetramers have a dimer-of-dimers topology, and individual subunits within the human tetramerization domain (residues 325–355) are composed of a short β-strand followed by a sharp turn leading into an α helix.
After the discovery of the p53 gene in humans, monkeys, chickens, and mice, it was mapped. The human p53 gene was located on 17p13, the mouse p53 was located on chromosome 11 and a nonfunctional pseudogene was found on chromosome 14. Among animals with different degrees of evolution, p53 has a very similar gene structure, about 20 KB long, composed of 11 exons and 10 introns. The first exon is non-coded, and exon-2, -4, -5, -7, and -8 encode five evolutionarily highly conserved domains and five highly conserved regions of the p53 gene, namely the 13-19, 117-142, 171-192, 236-258, and 270-286 coding regions. The p53 gene is further transcribed into a 2.5 kb mRNA, encoding 393 amino acid proteins with a molecular weight of 43.7 kDa. The expression of the p53 gene is regulated at least at the transcriptional and post-transcriptional levels.
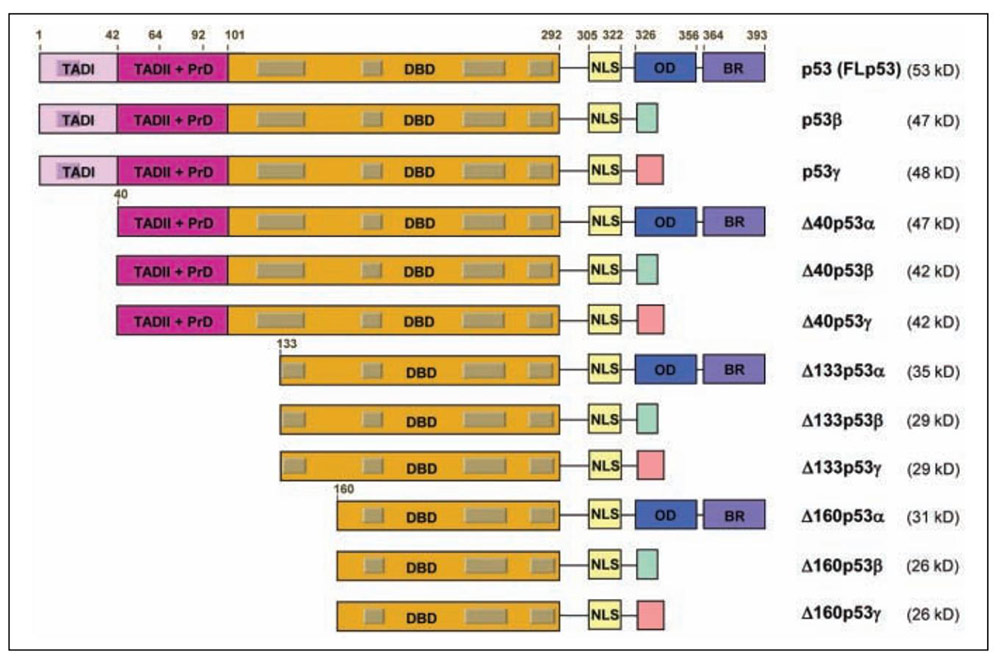
Fig1. The human p53 gene expresses 12 distinct p53 protein isoforms. (Marie P Khoury, 2011)
The p53 Pathway Activation
The p53 pathway activation is a critical cellular response to various stress signals, including DNA damage, oxidative stress, and oncogene activation. The p53 protein, a nuclear transcription factor, plays a central role in this pathway by regulating the expression of numerous genes involved in apoptosis, cell cycle arrest, and senescence.
Activation of the p53 pathway can occur through multiple signaling mechanisms. For instance, DNA damage-induced activation of p53 involves the ataxia telangiectasia-mutated (ATM) kinase, which phosphorylates serine residues in the p53 protein, leading to its stabilization and activation. Additionally, oxidative stress can activate p53 through the c-Jun N-terminal kinase (JNK) signaling pathway, which is independent of the DNA damage response (DDR).
The p53 pathway is also regulated by various upstream signals, including the PI3K/Akt pathway. Under normal conditions, activation of Akt can lead to the inhibition and degradation of p53. However, under stress conditions, such as DNA damage, the PI3K/Akt pathway is inhibited, allowing for the activation of p53. This highlights the complex interplay between signaling pathways in the regulation of p53 activity.
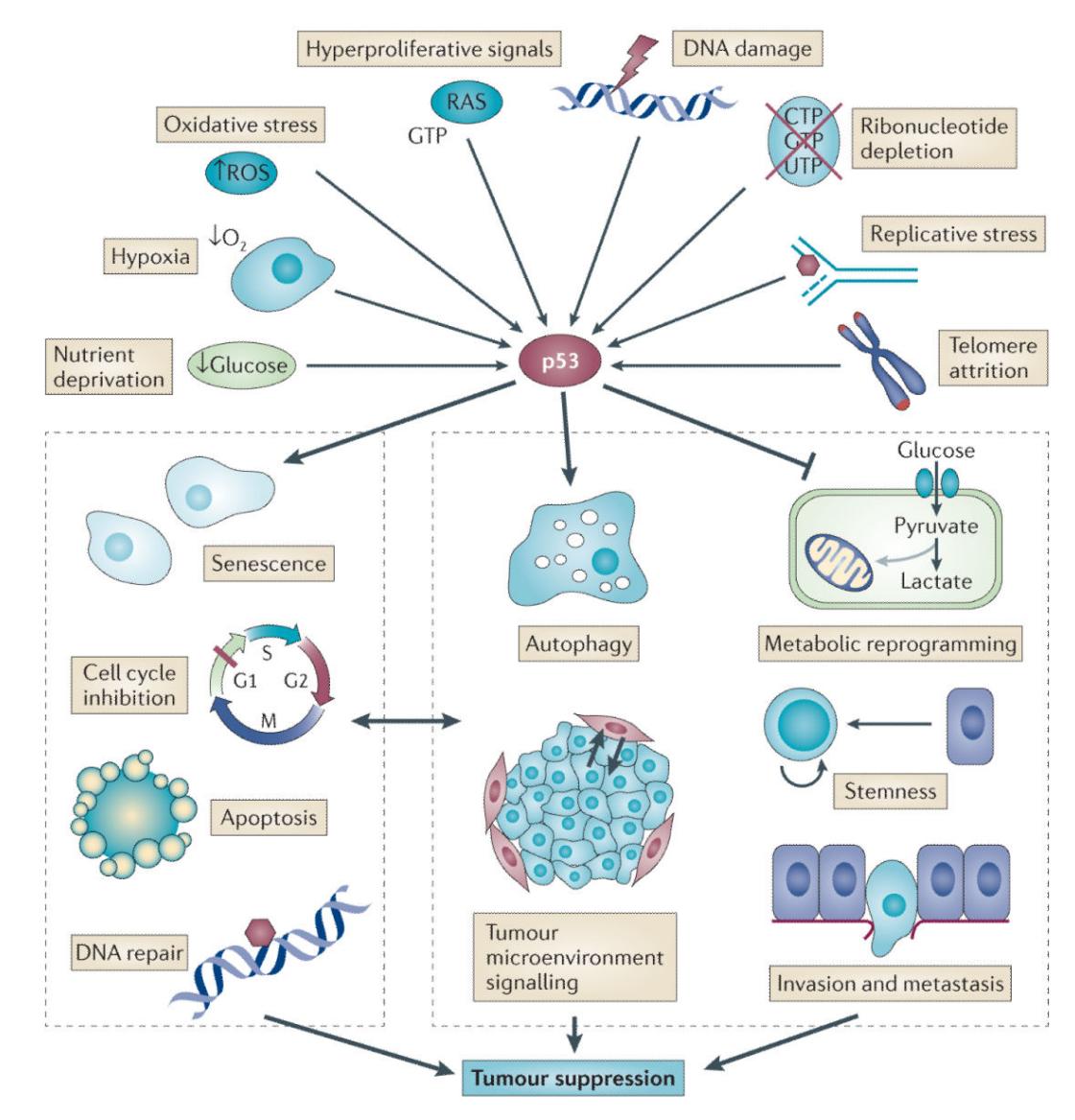
Fig2. A revised view of p53-activating signals and responses that are important for tumour suppression. (Kathryn T Bieging, 2014)
Downstream Events of The p53 pathway
Cell-cycle arrest: Cell-cycle arrest by p53 is mainly mediated by the transcriptional activation of p21/WAF1. p53 can activate the transcriptional upregulation of CDKN1A, which encodes for cell cycle inhibitor p21. The p21 mRNA is highly induced after p53 activation and is the first p53 target gene. p21 binds to cyclin E/Cdk2 and cyclin D/Cdk4 complexes to cause G1 arrest in the cell cycle. In addition, p53 activation also arrests cells at the G2/M phases. Although p21 can also inhibit cyclin B/Cdc2 from inhibiting cell-cycle progression through mitosis, other p53 target genes, such as 14-3-3σ, may participate in blocking the G2/M transition.
Cellular senescence: Cellular senescence mediates via p53-induced transcriptional activation of the cyclin-dependent kinase (CDK) inhibitors P21CIP1 (CDKN1A) and P16 INK4A (CDKN2A). The role of P21CIP1 may be to initiate senescence, whereas P16INK4A may be responsible for durable growth arrest. Apart from P21INK4A, there are other target genes like PML nuclear body scaffold (PML) and SERPINE1 (Serpin family E member 1), also known as Plasminogen activator inhibitor-1 (PAI-1), which are transcriptionally activated by p53 and plays a role in senescence.
Apoptosis: Many apoptosis-related genes transcriptionally regulated by p53 have been identified, which are candidates for implementing p53 effector functions. In response to oncogene activation, p53 mediates apoptosis through a linear pathway involving Bax transactivation, Bax translocation from the cytosol to membranes, cytochrome c release from mitochondria, and caspase-9 activation, followed by the activation of caspase-3, -6, and -7. p53-mediated apoptosis can be blocked at multiple death checkpoints by inhibiting p53 activity directly, by Bcl-2 family members regulating mitochondrial function, by E1B 19K blocking caspase-9 activation, and by caspase inhibitors.
p53 Related Diseases
p53 is a crucial tumor suppressor protein involved in preventing cancer development by responding to various cellular stresses, such as DNA damage, oxidative stress, and oncogene activation. Mutations in the TP53 gene, which encodes the p53 protein, are associated with a wide range of human diseases, most notably various types of cancer.
Cancer: TP53 mutations are found in over 50% of all human cancers, making it one of the most frequently mutated genes in cancer. These mutations often result in a loss of p53's tumor suppressor function, leading to uncontrolled cell growth and the development of tumors. Notably, TP53 mutations are particularly prevalent in cancers such as ovarian, colorectal, esophageal, and lung cancer, among others.
Leukemia: TP53 mutations are also associated with the development of certain types of leukemia, including acute myeloid leukemia (AML). Chronic inflammation, which is a common feature of leukemia, has been identified as a driver of TP53-mutant leukemic evolution, suggesting that TP53 mutations play a key role in the evolution of leukemia under inflammatory conditions.
Immune System Disorders: Recent research has highlighted the role of p53 in the regulation of the immune system. Mutations in TP53 can affect the immune response to cancer cells, with potential implications for the effectiveness of immunotherapies. For example, wild-type p53 can enhance the immunogenicity of cancer cells, while mutant p53 may impair the immune system's ability to eliminate cancer cells.
Li-Fraumeni Syndrome: This rare inherited condition is characterized by a predisposition to early onset of sarcomas and breast cancers. It is caused by germline mutations in the TP53 gene, which means the mutation is present in every cell of the body from birth. Individuals with Li-Fraumeni Syndrome have a significantly increased risk of developing multiple cancers throughout their lifetime.
Hereditary Nonpolyposis Colorectal Cancer (HNPCC): Also known as Lynch syndrome, this is an inherited condition that increases the risk of colorectal cancer. While TP53 mutations are not the primary cause of HNPCC, individuals with TP53 mutations may have an increased risk of developing colorectal cancer in addition to other types of cancers.
Case Study
Case Study 1: Recombinant Human TP53 protein (TP53-185H)
p53 plays an important role in the safeguard of the genome but it is frequently downregulated mainly by E3 ubiquitin ligases among which COP1 plays an important role. The overexpression of COP1 has been reported to occur in several tumors and may be indicative of its overall oncogenic effect, which in turn might be originated by a direct interaction of COP1 with p53. Such an interaction may constitute a rewarding target for anticancer drug design strategies; therefore, a deeper understanding of its underlying molecular mechanism and kinetics is needed. The formation of a single p53-COP1 bimolecular complex was visualized by atomic force microscopy imaging on a mica substrate. The kinetic characterization of the complex, performed by atomic force spectroscopy and surface plasmon resonance, provided a KD value of ∼10-8 M and a relative long lifetime in the order of minutes, both at the single-molecule level and in bulk solution. The surprisingly high affinity value and low dissociation rate of the p53-COP1 bimolecular complex, which is even stronger than the p53-MDM2 complex.
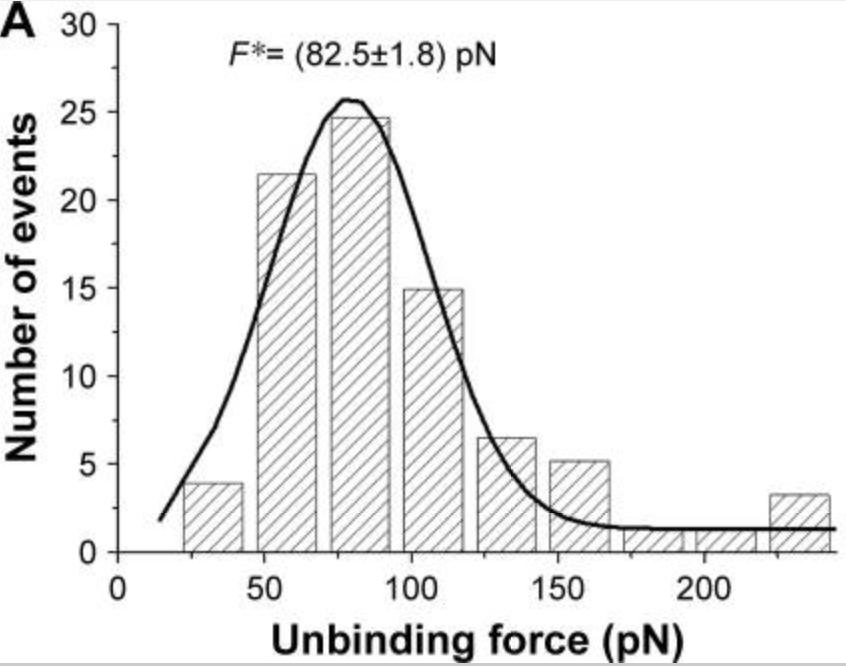
Fig1. Histogram of the unbinding forces for the p53–COP1 complex from AFS measurements carried out at a loading rate of 7 nN/s. (Ilaria Moscetti, 2018)
Case Study 2: Recombinant Human CASP3 (CASP3-158H)
Proteolytic truncation of microtubule associated human (h) Tau protein by caspase-3 at the carboxy (C) terminus has been linked to the pathogenesis of Alzheimer's Disease (AD). Interestingly, it is further noted that Ser422 residue present in the P1' position of hTau caspase-3 cleavage region is a potential phosphorylation site. The goal of this project is to study in vitro the caspase-3 cleavage site of hTau protein and to examine the kinetics of this cleavage following Ser422 phosphorylation and treatment with caspase-3 inhibitors. Peptides were designed from 441-mer major human Tau protein sequence that encompasses the proposed caspase-3 cleavage site [Asp421↓Ser422]. Corresponding phospho-, dextro-Ser422 and dextro-Asp421 analogs were also designed. These peptides were then incubated with recombinant caspase-3 enzyme under identical condition for digestion and analyzed for cleavage by mass spectrometry and RP-HPLC chromatograms. The results indicated that while the control peptide is efficiently cleaved by caspase-3 at Asp421↓Ser422 site producing the expected N- and C-terminal fragment peptides, the corresponding phospho-Ser422 peptide remained completely resistant to the cleavage. Substitution of Asp421 by its dextro isoform also blocks peptide cleavage by caspase-3. However substitution of Ser422 by its dextro isoform in the peptide did not affect the cleavage significantly.
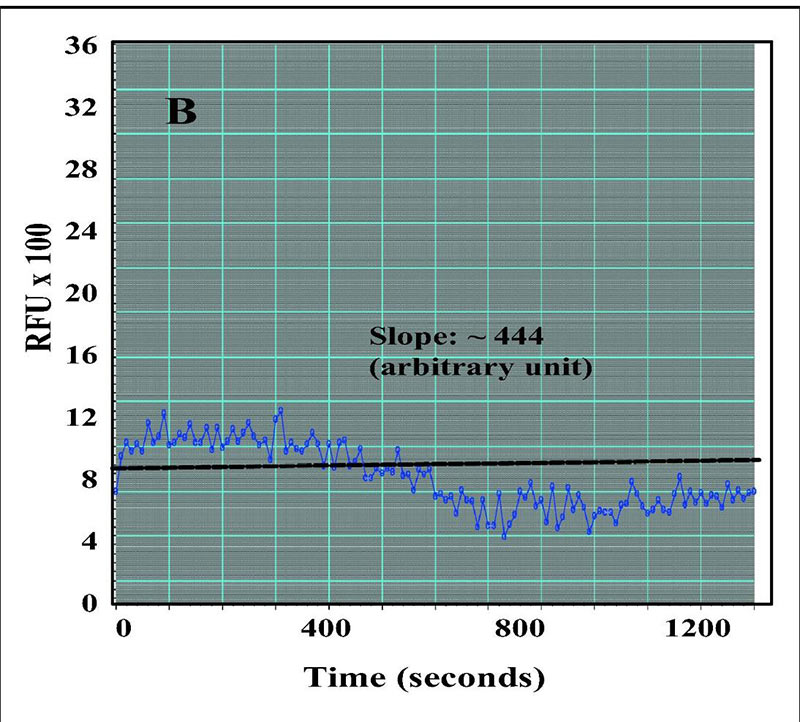
Fig2. In vitro caspase-3 activity assay in presence of inhibitor Ac-DQVD-aldehyde. (Priya Sandhu, 2017)
Case Study 3: Recombinant Human BCL2
Spinal cord injury (SCI) can result in structural and functional damage to the spinal cord, which may lead to loss of limb movement and sensation, loss of bowel and bladder control, and other complications. Previous studies have revealed the critical influence of trans-acting transcription factor 1 (SP1) in neurological pathologies, however, its role and mechanism in SCI have not been fully studied. The study was performed using mouse microglia BV2 stimulated using lipopolysaccharide (LPS) and male adult mice subjected to spinal hitting. Western blotting was performed to detect protein expression of SP1, 5-hydroxytryptamine (serotonin) receptor 2B (HTR2B), BCL2-associated x protein (Bax), B-cell lymphoma-2 (Bcl-2). Cell viability and apoptosis were analyzed by MTT assay and TUNEL assay. The association of SP1 and HTR2B was identified by chromatin immunoprecipitation assay and dual-luciferase reporter assay. The results showed that LPS treatment induced cell apoptosis and inhibited microglia polarization from M1 to M2 phenotype, accompanied by an increase of Bax protein expression and a decrease of Bcl-2 protein expression, however, these effects were relieved after SP1 silencing. SCI mouse model assay further showed that SP1 downregulation could attenuate spinal hitting-induced promoting effects on cell apoptosis of spinal cord tissues and microglial M1 polarization.
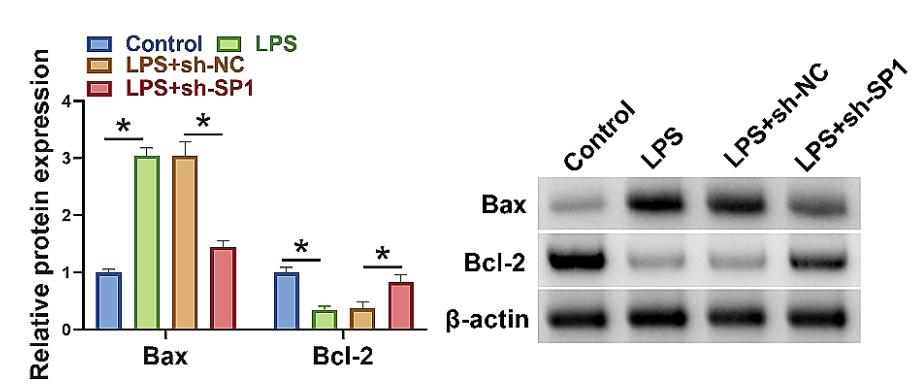
Fig3. Western blotting assay was used to detect protein expression of Bax and Bcl-2. (Qifei Xu, 2024)
Related Products
Despite nearly 40 years of research on p53, resulting in a staggering 80,000 publications, there are only beginning to grasp the full complexity of the p53 pathway, and the future will undoubtedly bring many novels, sometimes surprising, or even paradigm-shifting discoveries. Nevertheless, Creative BioMart is ready to provide a list of related protein products to support your ongoing research. Please feel free to contact us if you’re interested.
