


Protocol of Preparation of the Subcellular Organelle Protein Samples
In organism, the compartmentalization of cell makes different subcellular organelles have their own protein system, and many functions of protein can be realized through subcellular organelles. In addition, organelles have dynamic structures, such as endoplasmic reticulum, nucleus and Golgi apparatus. In addition to resident proteins, they also contain shuttle proteins and proteins that interact instantaneously. The research on these components, namely the subcellular organelle proteome research, will help to comprehensively excavate these proteins, so as to further elaborate the functions of organelles. Therefore, it is necessary to isolate subcellular organelles and study the expression of subcellular proteins. Mastering the specificity of these proteins will help to understand their specific functions and further elaborate the functions of organelles. The static subcellular localization analysis of the endomembrane system can obtain the localization information of subcellular proteins, which is helpful to further understand the subcellular proteome.
With the development of molecular biology, molecular genetics, genetic engineering and other disciplines in recent years, the research work on biological macromolecules contained in various organelles (such as nucleic acids, and proteins) is increasing day by day. Isolation of various specific proteins on various organelles has become one of the important contents of the preparation of biological macromolecules.
The subcellular organelles protein samples are actually prepared by pre-separation of protein. Generally, the organelles are separated by differential centrifugation or other methods, and then the protein in these specific organelles or proteins with specific properties are obtained.
Master the main methods and corresponding precautions for the separation of animal and plant organelles, master the preparation methods of subcellular protein samples and understand the importance of subcellular proteomics.
When preparing certain biological macromolecules according to experimental needs, it is usually necessary to use specific parts of cells as materials. In order to purify biomacromolecules on specific organelles, usually after breaking cells, the organelles are separated first, and then the required biomacromolecule components are obtained by corresponding methods.
The organelles are usually separated by differential centrifugation. After the cells are broken, differential centrifugation is carried out in an appropriate medium. each component of cells with different mass and size will settle in different areas of the centrifuge tube, and the required components are obtained after separation. The selection of medium is very important during the separation and preparation of organelles. The first medium used was normal saline, which is easy to make the sub-organelle particles aggregate and form blocks. Consequently, sucrose, Ficoll (a sucrose polymer) or glucose polyethylene glycol and other polymer solutions are generally used instead. The purity of the isolated organelles can be identified by electron microscopy, immunochemistry or determination of marker enzyme activity.
The distribution of various biological macromolecules in cells is different. Almost all DNA is concentrated in the nucleus. RNA is mostly distributed in the cytoplasm. The distribution of various enzymes in cells also has a certain position. Therefore, when preparing biomacromolecules on organelles, it is necessary to know the whole cell structure and the distribution properties of various biomacromolecules in cells in advance (see Figure 1-2-1 for the sedimentation coefficients of different molecules and organelles; see Table 1-2-1 for the size and precipitation characteristics of organelles)
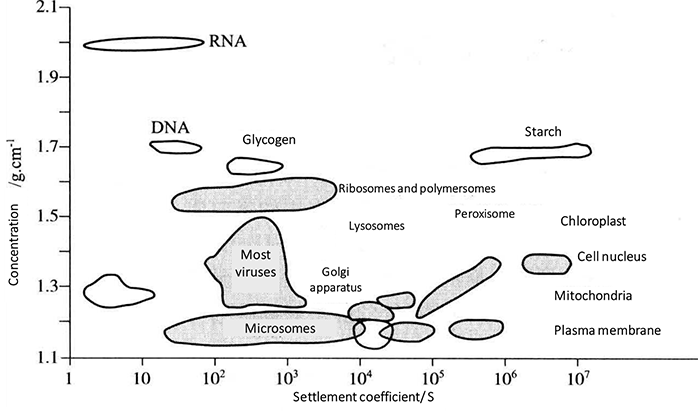 Figure 1-2-1 Sedimentation Coefficient of Different Molecules and Organelles
Figure 1-2-1 Sedimentation Coefficient of Different Molecules and Organelles
Table 1-2-1 Size and Precipitation Characteristics of Organelles
| Subcellular organelle | Size /μm | Centrifugal force/g *1 | Time /min |
|---|---|---|---|
| Nucleus | 4-12 | 500-1000 | 5-10 |
| Nuclear membrane | 2000 (30000) | 30(5) | |
| Mitochondrion | 0.4-2.5 | 1000-10000 | 10-15 |
| Lysosome | 0.4-0.8 | 6000-15000 | 10-20 |
| Peroxisome | 0.4-0.8 | 6000-15000 | 10-20 |
| Rough endoplasmic reticulum vesicles | 0.05-0.35 | 30000-100000 | 30-60 |
| Smooth endoplasmic reticulum vesicles | 0.05-0.3 | 50000-100000 | 30-60 |
| Large cell membrane | 3-20 | 1000-3000 | 10-15 |
| Vesicle | 0.05-2.0 | 50000-100000 | 30-60 |
| Nuclear endosome | 0.05-0.4 | 50000-100000 | 30-60 |
| Golgi apparatus (complete) | 1.0-2.0 | 10000-20000 | 20-30 |
| Golgi apparatus (vesicles) | 0.05-0.5 | 50000-100000 | 20-40 |
| Sarcoplasmic reticulum | 0.1-1.0 | 10000-35000 | 20 |
| Chloroplast | 2-5 | 1000-2000 | 10 |
| Plant mitochondria | 1-3 | 5000-20000 | 15 |
1. Cell Fragmentation
For the first step of separating subcellular components, please refer to the content of cell fragmentation in Module 1-1. The problem to be noted is that in this part of the experiment, more gentle fragmentation methods are used, and microscopic monitoring can be used to ensure the structural integrity of organelles. The method of preparing tissue homogenate or cell homogenate is usually used in the experiment. Homogenization refers to putting tissues or cells into a homogenizer and adding isotonic homogenizing medium (i.e., 0.25 mol/L sucrose and 0.003 mol/L calcium chloride solution), so that cells are mechanically crushed into a mixture of various subcellular components and contents.
2. Separation of Subcellular Components
The separation of subcellular components needs to adopt the hierarchical separation method, which mainly uses the centrifugation technology from low speed to high speed to make the particles in the heterogeneous mixture settle to different parts of the centrifuge tube in batches according to their size and density, and then collect them separately to obtain various subcellular components. Since particles of different sizes and densities in the sample are uniformly distributed in the whole centrifuge tube at the beginning of centrifugation, the first precipitation obtained by each stage of separation must not be the pure and heaviest particles, and must be purified by repeated suspension and centrifugation. The main centrifugation techniques for separating subcellular components are differential centrifugation and density gradient centrifugation.
(1) Differential Centrifugation
Centrifuge step by step from low speed to high speed in a medium with uniform density to separate cells and organelles of different sizes. In differential centrifugation, the sequence of organelle sedimentation is generally as follows: nucleus, mitochondria, lysosome and peroxisome, Golgi apparatus and endoplasmic reticulum, and finally ribosome. Because various organelles overlap each other in size and density, and some slow settling particles are often wrapped in the settling block by fast settling particles, it generally needs to repeat 2-3 times to obtain ideal results. The organelles preliminarily separated by differential centrifugation often need to be further separated and purified by density gradient centrifugation. The principle of differential centrifugation is shown in Figure 1-2-2. See Table 1-2-2 and Table 1-2-3 for the contents of precipitates formed by differential centrifugation of plant materials and liver materials.
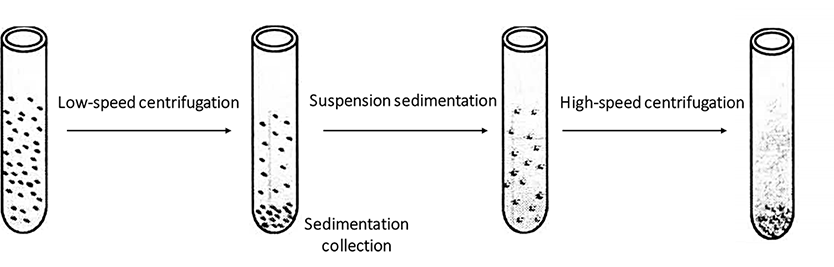 Figure 1-2-2 The Principle of Differential Centrifugation (the Centrifugation Speed is Gradually Increased, and the Samples are Precipitated Successively from Large to Small)
Figure 1-2-2 The Principle of Differential Centrifugation (the Centrifugation Speed is Gradually Increased, and the Samples are Precipitated Successively from Large to Small)
Table 1-2-2 Content of the Precipitate (Plant) Obtained by Differential Centrifugation
| Sedimentation | (Centrifugal force/g) X (Time/min) | Contents |
|---|---|---|
| A | 150 X 20 | Intact cell |
| B | 1000 X 20 | Nucleus, cell debris |
| C | 3000 X 6 | Chloroplast |
| D | 10000 X 20 | Mitochondria, lysosomes, microsomes |
| E | 105000 X 120 | Microbody |
| F | 105000 X 20 + 0. 26% Sodium deoxycholate | Ribosome |
Table 1-2-3 Content of the Precipitate (Liver Materials) Obtained by Differential Centrifugation
| Sedimentation | (Centrifugal force/g) X (Time/min) | Contents |
|---|---|---|
| P1 | 1000 X 10 | Nucleus, heavy mitochondria, large cell membranes |
| P2 | 3000 X 10 | Heavy mitochondria, cell membrane fragments |
| P3 | 6000 X 10 | Mitochondria, lysosomes, peroxisomes, intact Golgi bodies |
| P4 | 10000 X 10 | Mitochondria, lysosomes, peroxisomes, Golgi membranes |
| P5 | 20000 X 10 | Lysosomes, peroxisomes, Golgi membrane, large high-density vesicles (such as rough endoplasmic reticulum) |
| P6 | 100000 X 10 | All vesicles, cell membranes, Golgi bodies, nuclear endosomes, etc. from the endoplasmic reticulum |
(2) Density Gradient Centrifugation
Use a certain medium to form a continuous or discontinuous density gradient in the centrifuge tube, place the cell suspension or homogenate on the top of the medium, and delaminate and separate the cells by gravity or centrifugal force. Density gradient centrifugation can also be divided into velocity sedimentation and isopycnic sedimentation (Table 1-2-4). See Figure 1-2-3 for the process of velocity sedimentation and isodensity sedimentation.
Table 1-2-4 Classification of Density Gradient Centrifugation
| Velocity sedimentation | Isopycnic sedimentation | |
|---|---|---|
| Separation type | It can be used to separate biological particles (cells or organelles) with similar density but different sizes. | It can be used to separate particles with different densities. It is suitable for cell isolation, but not suitable for cell isolation and purification. |
| Media density | Low medium density. Maximum density of medium < minimum density of separated particles. |
The medium density is high. Maximum density of medium > maximum density of separated components. |
| Separation process | Settle and separate at different speeds in a very gentle density gradient medium according to their respective settlement coefficients. | In the medium with continuous density gradient, the large centrifugal force will settle to the medium with the same density and complete the separation. |
| Advantages and disadvantages | The required force field is 10-100 times larger than the velocity sedimentation. High speed or ultracentrifugation is often required, and the time is long, which is unfavorable to cells. |
Generally, differential centrifugation and density gradient centrifugation are used together. The cell lysates produced by homogenate are separated from organelles and other particles by a series of differential centrifugation steps and density gradient centrifugation. Different organelles were separated by different methods (Table 1-2-5).
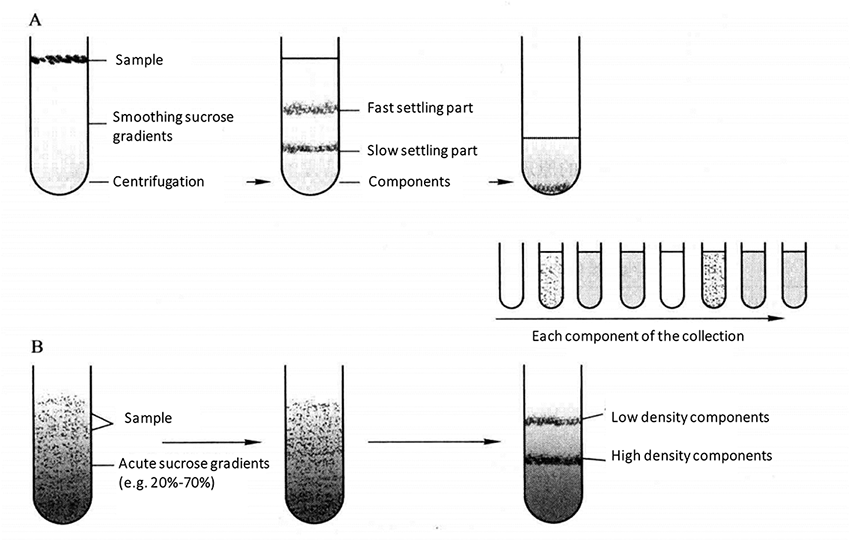 Figure 1-2-3 A. Velocity Sedimentation; B. Isopycnic Sedimentation
Figure 1-2-3 A. Velocity Sedimentation; B. Isopycnic Sedimentation
Table 1-2-5 Separation Methods Chosen for Specific Organelles
| Organelles to be separated | Separation method |
|---|---|
| Cell membrane (erythrocyte membrane, mitochondrial membrane, other cell membranes) | Differential centrifugation |
| Mitochondrion | Gradient centrifugation |
| Mitochondrial membrane | Density gradient centrifugation *2 |
| Polyribosome | Differential centrifugation |
| Microsome | Fractional centrifugation. Ultracentrifugation after removal of nucleus and mitochondria |
| Nucleus | Fractional centrifugation |
3. Analysis and Confirmation of Subcellular Components
The third step of separating subcellular components is to analyze and confirm the separated components. Common analysis methods include identification of morphology and function.
1. Main Instruments and Equipment
High-speed centrifuge, ultracentrifuge, ultrasonic crusher.
2. Experimental Materials
Cultured cells, plant or animal tissues.
3. Main reagents
(1) Homogenate*3
| 0.25 mol/L | Sucrose |
| 0.003 mol/L | Calcium chloride |
(2) Homogenization Buffer
| 0.38 mol/L | Mannitol |
| 4-50 mmol/L | Tris-HCl (pH 8.0) |
| 1 mmol/L | DDT |
| 0.05% | Bovine serum albumin (BSA) |
(3) Isotonic Solution Buffer A
| 250 mmol/L | Mannitol |
| 75 mmol/L | Sucrose |
| 0.1 mmol/L | EDTA |
| 0.5 mmol/L | EGTA |
| 1 mmol/L | PMSF |
| 10 mmol/L | HEPES (pH 7.4) |
(4) Isotonic Solution Buffer B (Buffer A + 0. 5% BSA Media Solution)
| 0. 15 mol/L | Sucrose |
| 0. 025 mol/L | KCl |
| 0. 1 mol/L | Tris-HCl (pH 7.4) |
| 0. 005 mol/L | MgCl2 |
(5) Suspensions
| 0.33 mol/L | Sorbitol |
| 20 mmol/L | Tris-NaOH (pH 8.4) |
| 10 mmol/L | EDTA |
| 0.17 mol/L | NaCl |
| 1 % | BSA |
(6) Lysis Solution
| 50 mmol/L | Tris |
| 0. 1 % Triton | X-100 |
| 100 mmol/L | KCl |
| 20% | Glycerin (pH 7.0) |
(7) Lysis Solution 1
| 8 mol/L | Urea |
| 40 mmol/L | Tris-base |
| 4% | CHAPS |
| 65 mmol/L | DTT |
(8) Lysis Solution 2
| 8 mol/L | Urea |
| 2 mol/L | Thiourea |
| 50 mmol/L | Tris |
| 0. 2 mol/L | KCl |
| 20 % | Glycerin |
| 10 % | SDS |
| 0.2 % | Triton X-100 |
(9) Lysis Solution 3
| 50 mmol/L | Tris |
| 1 mmol/L | MgSO4 |
Example 1. Separation of Nucleus
(1) Kill mice by decapitation and take the liver by laparotomy. Cut the liver into small pieces (remove connective tissue) and place it in a beaker containing 0.9% NaCl as soon as possible. The liver should be washed repeatedly to remove blood stains as much as possible. Suck the liquid on the surface of liver with filter paper.
(2) Put the liver tissue with a wet weight of about 1g into a small plate, measure 8 mL of precooled homogenate A with a measuring cylinder, add a small amount of this solution into the plate, cut the liver tissue as much as possible, and then add all of homogenate A.
(3) The cut liver tissue is poured into the homogenizing tube, homogenized for 3-5 times under the ice bath condition, and filter the homogenizing solution into the centrifuge tube with three layers of gauze.
(4) Homogenization solution 2500 r/min, centrifugation for 15min. Take the supernatant liquid*4 and transfer it to a high-speed centrifuge tube, and store it in a beaker with ice cubes for separation of mitochondria. Proceed to the next step with the sediment.
(5) Suspend the sediment with 6 mL homogenate A, 2500r/min, centrifuge for 15min, and discard the supernatant. Blow the residual liquid into suspension with a straw, which is the nucleus obtained from separation.
Example 2. Isolation of Mitochondria and Protein Extraction
(1) Kill the mice by decapitation and take the liver immediately.
(2) Put 1 g of liver tissue into 100 mL ice bath isotonic solution Buffer A, homogenized 5-8 times. Centrifuge the homogenized solution 1000 g 10 min twice to remove the cell membrane and nuclear components.
(3) Take the supernatant, centrifuge 10 000 g for 15 min.
(4) The precipitate obtained by centrifugation was the mitochondrial component of the liver tissue. Use 10 mL of isotonic solution Buffer B (Buffer A + 0 5% BSA) wash the precipitate twice, add 1 mL of lysate to lyse, and then conduct 100 W 4 X 15s ultrasound.
(5) Centrifuge 25 000 g at 4 ° C for 1 h, and the supernatant is the prepared mitochondrial protein*5.
(6) Centrifuge the supernatant part for 1h with 100000 g, precipitated and enriched the microcomponent protein, and then cracked with 1 mL of lysate. The treatment process to get microcomponent protein was the same as that of mitochondrial components*6.
Example 3. Isolation of Microsomes*7
(1) The rat, weighing about 300g, was decapitated after 20 hours of starvation. The liver was taken out, washed, cut into pieces, added with a medium solution twice the volume, and homogenized in a glass homogenizer.
(2) Centrifuge 15000 g for 10 min, discard the sediment and take the supernatant.
(3) 100000 g centrifugation for 60 min, discard the supernatant, take the sediment, and the sediment is the microsome.
(4) Store microsome in medium solution.
Example 4. Isolation of Lysosomes (all the following operations must be performed at 0-4 °C)
(1) Prepare sucrose gradient solution. Take a gradient mixer with two small cups, and load 117 mL of sucrose (2.1mol/L) and 13mL of sucrose (1.1mol/L) into the two small cups respectively.
(2) The rats were killed and the kidneys were taken out, 0.3mol/L sucrose was added in a ratio of 1:8 (W/V), and then the kidney tissues were homogenized in a glass homogenizer.
(3) Centrifuge 150g for 10min, discard the sediment, take the supernatant, centrifuge 9000g for 3min, discard the supernatant, and take the sediment.
(4) From top to bottom, the precipitates are white, yellowish brown and dark brown in order. The upper layer is a mixture of membrane components, the middle layer is a mitochondrial part, and the bottom layer is a semi purified lysosome *8.
(5) Carefully suck out the upper layer with a straw, then add a few milliliters of 0.3mol/L sucrose along the pipe wall, slowly shake the straw to suspend the interface layer, and discard the interface layer. Wash with 0.3mol/L sucrose once, and the bottom lysosome part is suspended in 2.5mL0.3mol/L sucrose.
(6) Lay 2mL suspended semi-purified lysosome on the sucrose gradient, stir the top gradient with a glass rod to destroy the interface between the sucrose gradient and lysosome, and then centrifugate 100000g for 150min. The centrifugation results show that the sucrose gradient solution is divided into three distinct bands and less precipitation. The dark yellow to brown band at the bottom is the purified lysosome.
Example 5. Chloroplast Isolation and Protein Extraction 1 *9
(1) Wash the materials, drain the water with filter paper, remove the veins, weigh 5g fresh weight leaves, and cut them into pieces.
(2) Put the fresh leaves into the mortar, measure 25mL of homogenate buffer solution (containing 0.05% bovine serum albumin *10), first pour 15-20mL into the mortar, then add a little quartz sand to fully grind *11.
(3) Filter the grinding solution into a 50mL centrifuge tube through 8 layers of gauze, wash the remaining homogenate into a mortar, and then filter it into a tube for 1000g centrifugation for 20min.
(4) The filtrate is centrifuged at 3000g for 6min.
(5) The precipitate was again suspended in 10mL homogenate buffer, centrifuged at 3000g for 6min, and the precipitate was purified chloroplast *12.
Example 6. Chloroplast Isolation and Protein Extraction 2 *9
(1) Take 100g spinach functional leaf *13, wash it with precooled distilled water and dry it with absorbent paper.
(2) After grinding with liquid nitrogen, add 120mL suspension and mix well.
(3) The suspension was filtered by multi-layer gauze. The filtrate was first centrifuged at 4 °C for 10 min after 500g, and the complete cells and nuclei were removed. The supernatant was retained.
(4) After 1500g centrifugation at 4 °C for 15min, discard the supernatant, transfer the precipitation to a 50mL centrifuge tube, and mix it with 60% sucrose, so that the final concentration of the mixture is 55%.
(5) Slowly add 35% sucrose solution to the top to form a sucrose density gradient. Centrifuge at 4 °C for 7000g for 1h. The complete chloroplast will appear in the green layer between the two layers of the sucrose density gradient of 35%~55%. Carefully absorb the upper layer of sucrose solution, transfer the chloroplast to another 50mL tube, and resuspension it with suspension medium.
(6) At 4 °C, centrifugate and wash 7000g for 30min twice to remove impurities such as sucrose, and the precipitate is chloroplast *14.
(7) Extract hydrophilic protein *15 from the matrix with lysate 1.
(8) After being treated at 4 °C for 1h, 14000g of the supernatant was centrifuged for 40min.
(9) The precipitates were treated with lysate 2 to obtain hydrophilic, neutral and hydrophobic proteins in the organelles.
(10) After being treated at 4 °C for 1h, centrifuge it at 4000g for 40min, keep the supernatant.
(11) Treat the insoluble components (mainly basic proteins) in (2) are with lysate 3.
(12) The supernatant obtained in each step above was treated with precooled acetone containing β-mercaptoethanol (V/V=1:9) at 4 °C for 1h, centrifuged at 14000g for 40min, and the precipitate obtained was the target protein.
1. The first step of subcellular organelle protein extraction is the extraction of organelles. It is necessary to formulate an extraction plan for different organelles. During the extraction process, attention should be paid to ensuring their integrity. At the end of the extraction, the integrity of organelles should be checked first.
2. Reagents introduced in the extraction of organelles may affect subsequent experiments such as protein extraction, which should be considered in the scheme design.
3. In the process of protein extraction, attention should be paid to the protection of proteins. Some proteins are easy to be degraded, so protease inhibitors and reducing agents can be added, and the operation should be kept at low temperature as far as possible.
*1 It is generally accepted that a centrifuge with a speed of 10,000-25,000 r/min is called a high-speed centrifuge. Centrifuges with a speed of more than 25,000 r/min and a centrifugal force of more than 89,000 g are called ultracentrifuges. At present, the maximum speed of ultracentrifuge can reach 100000 r/min and the centrifugal force is more than 500000 g.
*2 The common media for density gradient centrifugation are cesium chloride, sucrose and polysaccharose. The medium requirements for separating living cells: density gradient can be generated, and when the density is high, the viscosity is not strong; pH neutral or easily adjusted to neutral; when the concentration is large, the osmotic pressure is small; it is non-toxic to cells.
*3 The homogenate is mainly composed of sugar, such as mannitol and sucrose. Its main function is to maintain the osmotic pressure inside and outside the cells and ensure the complete morphology of cells.
*4 After the nuclei were separated, the supernatant and the precipitate suspension could be taken and dropped on the slide, and the nuclei were observed after staining with 1% toluidine blue.
*5 After the preparation of mitochondria, take the supernatant and sediment suspension, drop them on the slide, and dye them with 0.02% Janus Green B dye for 20 minutes. Under the oil microscope, the granular mitochondria were dyed blue-green by Janus Green B to judge the separation of mitochondria.
*6 When cells are broken by homogenate, the plasma membrane breaks into fragments, and the ends of these membrane fragments fuse to form vesicles with a diameter of less than 100 nm. Vesicles from different organelles (nucleus, mitochondria, plasma membrane, endoplasmic reticulum, etc.) have different characteristics, so they can be separated. Small vesicles derived from the intima system form vesicle heterogeneous aggregates of similar size, called microsomes.
*7 The method of microsome separation is to prepare microsome by ultracentrifugation after removing the nucleus and mitochondria by fractional centrifugation.
*8 Lysosomes are vesicular structures surrounded by a unit membrane and containing a variety of acid hydrolases. Lysosomes contain more than 40 kinds of hydrolases, including protease, nucleic acid degrading enzyme and glycosidase. Its main function is to digest substances in cells. In addition, lysosomes are closely related to organ formation, regulation of hormone secretion and the occurrence of some diseases.
*9 There are plastids in almost all plant tissues. Chloroplasts are the most thoroughly studied plastids.
*10 During homogenization, toxic substances stored in vacuoles will be released. In the species with the accumulation of phenolic compounds, the toxic effects can be reduced by adding soluble polyvinylpyrrolidone, bovine serum albumin (BSA) and mercapto compounds to the grinding buffer.
*11 Plants are wrapped in cell walls. The force of breaking cells should be enough to break the cell walls while maintaining the integrity of chloroplasts.
*12 The extraction method is improved according to the method of Maryse A and Walket.
*13 Due to the accumulation of starch into dense granules in chloroplasts, the chloroplasts will be broken during centrifugation. The accumulated starch can be eliminated by plants growing under the conditions of short light time and/or low light intensity. If necessary, plants can be put in the dark for 24-48h before homogenization to remove starch.
*14 Determination of chloroplast concentration. Take 50mL of chloroplast suspension, dissolve it in 20mL of acetone (80%), centrifugate to remove the precipitation, and compare the color of the supernatant at the wavelength of 652nm (optical path is 1cm) of the spectrophotometer. The measured optical density value multiplied by 100/g is the chlorophyll content in chloroplast suspension (mg/mL).
*15 Protein extraction and content determination (improved by Klose's fractional order method).