


IL-17 Family Receptors
Related Symbol Search List
Immunology Background
Available Resources for IL-17 Family Receptors Research
We are excited to introduce our wide array of products pertaining to IL-17 family receptors at Creative BioMart. Our product range includes premium recombinant proteins, protein pre-coupled magnetic beads, and cell and tissue lysates, ensuring that researchers have essential tools at their disposal. Understanding the unique nature of each research endeavor, we offer customizable services to cater to specific requirements, ensuring that the products align with your research goals.
Beyond our diverse product line, we are dedicated to providing a rich array of resources related to IL-17 family receptors. These resources cover various topics such as pathways, protein functions, interacting proteins, relevant articles, and research areas. Serving as a knowledge center, these invaluable resources empower researchers to enhance their comprehension of IL-17 family receptors and their pivotal roles in physiological processes.
Our Featured Products
| Cat.# | Product name | Species | Source (Host) | Tag |
|---|---|---|---|---|
| Il17ra-4026M | Active Recombinant Mouse Il17ra, His tagged | Mouse | HEK293 | His |
| IL17RC-14159H | Recombinant Human IL17RC, His-tagged | Human | E.coli | His |
| IL17RD-766H | Recombinant Human IL17RD protein(Met1-Arg299), His-tagged | Human | HEK293 | C-His |
| IL17RE-14161H | Recombinant Human IL17RE, GST-tagged | Human | E.coli | GST |
About IL-17 Family Receptors
The IL-17 family of cytokines exerts their effects by binding to specific receptors on target cells, and these receptors are collectively known as IL-17 family receptors. Each IL-17 family cytokine typically interacts with specific receptor complexes, which are composed of multiple subunits. Here's an introduction to the IL-17 family receptors:
Interleukin-17 receptor A (IL-17RA)
- IL-17RA is the primary receptor for IL-17A.
- It is widely expressed on various cell types, including epithelial cells, fibroblasts, and immune cells.
- IL-17A binds to IL-17RA, initiating downstream signaling pathways that promote inflammation and immune responses.
Interleukin-17 receptor C (IL-17RC)
- IL-17RC is a shared receptor subunit that can form heterodimeric receptor complexes with IL-17RA or IL-17RE.
- It is capable of binding IL-17A and IL-17F.
- The IL-17A/IL-17RC and IL-17F/IL-17RC complexes can activate similar downstream signaling pathways as IL-17A alone.
Interleukin-17 receptor B (IL-17RB)
- IL-17RB is the primary receptor for IL-17E (also known as IL-25).
- It is expressed on various cell types, including epithelial cells, smooth muscle cells, and immune cells.
- IL-17E binding to IL-17RB initiates signaling pathways that promote allergic responses, inflammation, and tissue remodeling.
Interleukin-17 receptor D (IL-17RD)
- IL-17RD is a receptor subunit that can form heterodimeric receptor complexes with IL-17RB or IL-17RE.
- It is involved in IL-17E signaling and amplifies the immune and inflammatory responses mediated by IL-17RB.
Interleukin-17 receptor E (IL-17RE)
- IL-17RE is a receptor subunit that can form heterodimeric receptor complexes with IL-17RA or IL-17RD.
- It is involved in IL-17F signaling and amplifies the immune and inflammatory responses mediated by IL-17F.
The binding of IL-17 family cytokines to their corresponding receptors triggers intracellular signaling cascades, leading to the activation of transcription factors and subsequent gene expression changes. These signaling pathways often involve the recruitment of adaptor molecules, such as Act1 (also known as CIKS), which play important roles in mediating downstream signaling events.
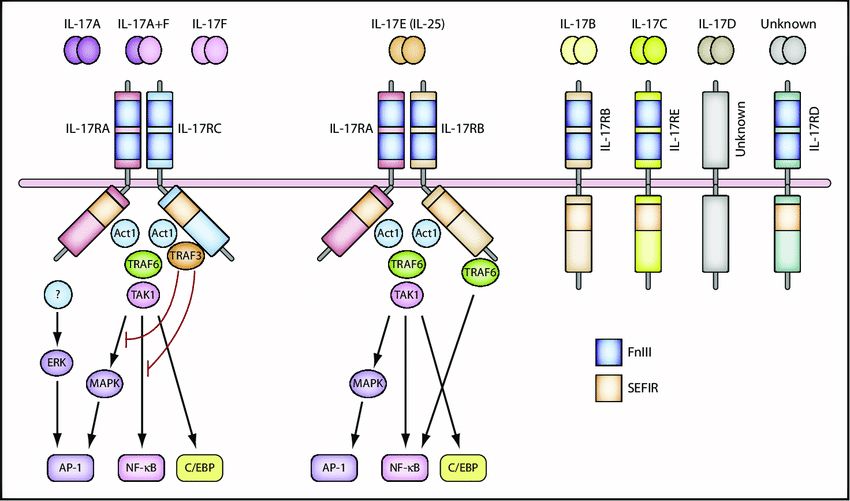 Fig.1 L-17 and the IL-17 receptor families. (Akitsu A, et al., 2012)
Fig.1 L-17 and the IL-17 receptor families. (Akitsu A, et al., 2012)
Role of IL-17 Family Receptors in Disease
IL-17 family receptors play a significant role in the development and progression of various diseases. Dysregulation of IL-17 receptor signaling has been implicated in autoimmune disorders, chronic inflammatory conditions, and certain cancers. Here are some examples of diseases where IL-17 family receptors are involved:
Autoimmune Diseases
IL-17 family receptors, particularly IL-17RA, IL-17RB, and IL-17RC, are implicated in several autoimmune diseases characterized by chronic inflammation and immune dysregulation. Conditions such as psoriasis, psoriatic arthritis, rheumatoid arthritis, ankylosing spondylitis, and systemic lupus erythematosus (SLE) involve aberrant IL-17 signaling. Activation of IL-17 receptors promotes the production of pro-inflammatory cytokines, chemokines, and tissue-destructive enzymes, contributing to disease pathology.
Inflammatory Bowel Disease (IBD)
IL-17 family receptors have been implicated in the pathogenesis of IBD, including Crohn's disease and ulcerative colitis. IL-17A and IL-17F, acting through their receptors, promote intestinal inflammation, recruitment of immune cells, and production of pro-inflammatory mediators in the gut. Blocking IL-17 receptor signaling has shown promise in reducing inflammation and improving symptoms in IBD patients.
Asthma
IL-17 family receptors play a role in the pathogenesis of severe asthma. IL-17A and IL-17F, acting through their receptors, contribute to airway inflammation, mucus production, and recruitment of neutrophils. Elevated IL-17 receptor signaling has been observed in the airways of asthmatic patients, and targeting IL-17 receptors has shown potential for asthma treatment.
Cancer
IL-17 family receptors are implicated in various cancers, particularly those associated with chronic inflammation. IL-17 receptor signaling can promote tumor growth, angiogenesis, and metastasis by stimulating the production of pro-inflammatory cytokines, chemokines, and growth factors. Additionally, IL-17 signaling may contribute to an immunosuppressive tumor microenvironment, hampering anti-tumor immune responses. Targeting IL-17 receptors or downstream signaling pathways is being explored as a potential therapeutic strategy in certain cancers.
It's important to note that the involvement of IL-17 family receptors in disease is context-dependent and can vary across different conditions. The specific contributions of IL-17 receptor signaling in each disease may involve complex interactions with other immune pathways and factors. Ongoing research continues to uncover the detailed mechanisms and potential therapeutic targets associated with IL-17 family receptor dysregulation in various diseases.
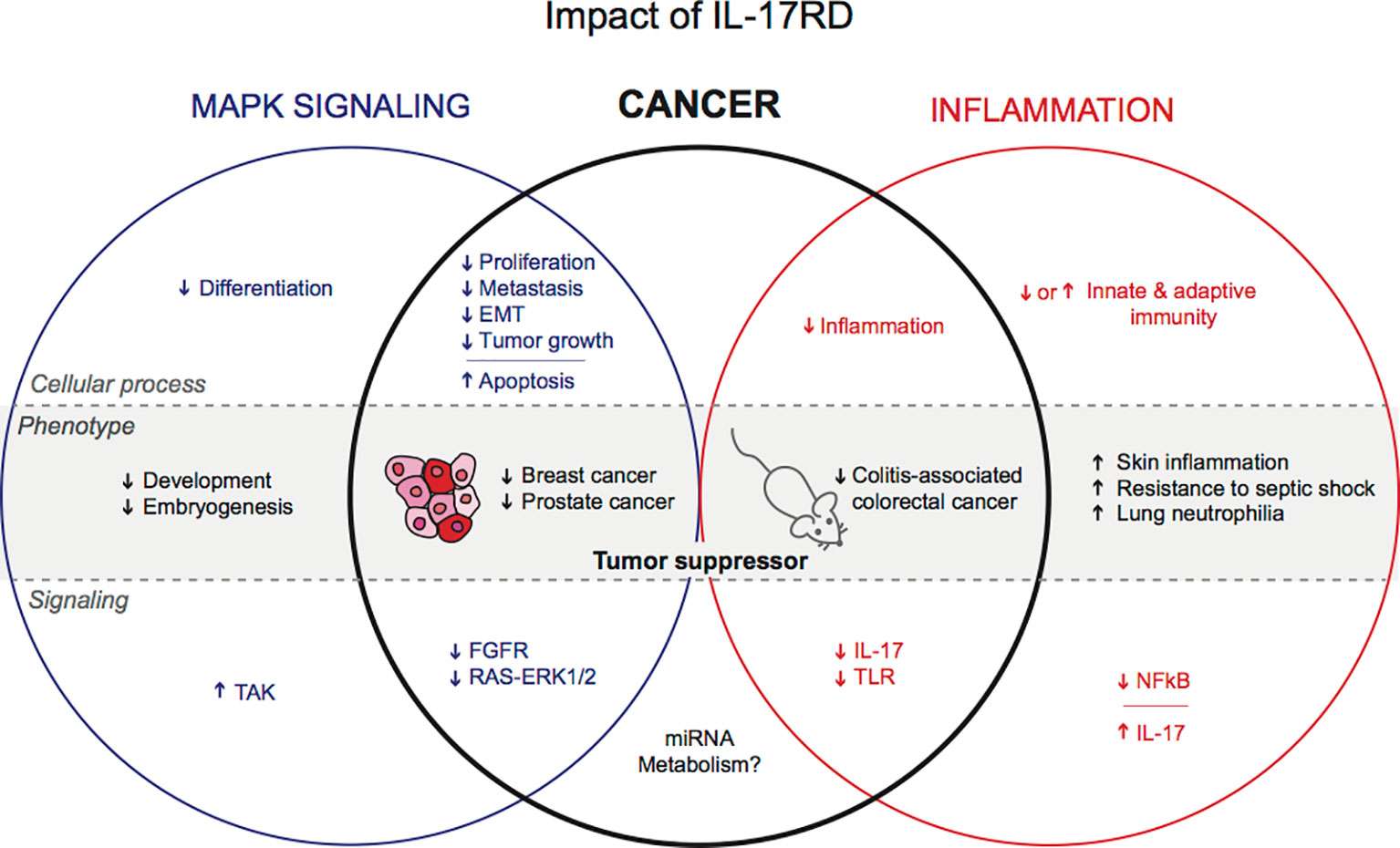 Fig.2 Postulated functions of IL-17RD in physiology and cancer. (Girondel C, et al., 2012)
Fig.2 Postulated functions of IL-17RD in physiology and cancer. (Girondel C, et al., 2012)
If you have any questions, requirements, or cooperation intentions, please feel free to contact us. We very much look forward to working with you and helping you achieve research and commercial success.
Related References:
- Akitsu A, Iwakura Y. Nihon Rinsho. IL-17 family and receptor pathway. 2012;70 Suppl 8:207-211.
- Girondel C, Meloche S. Interleukin-17 Receptor D in Physiology, Inflammation and Cancer. Front Oncol. 2021;11:656004. Published 2021 Mar 23. doi:10.3389/fonc.2021.656004