
IL-12 Family Receptors
Related Symbol Search List
Immunology Background
Available Resources for IL-12 Family Receptors Research
At Creative BioMart, we are delighted to present our extensive collection of products related to IL-12 family receptors. Our product portfolio comprises high-quality recombinant proteins, protein pre-coupled magnetic beads, and cell and tissue lysates, providing researchers with essential tools for their studies. We recognize that each research project is unique, which is why our customizable services are designed to meet your specific needs, guaranteeing that you receive the most suitable product tailored to your research goals.
In addition to our comprehensive product line, we are committed to providing a wealth of resources on IL-12 family receptors. Our resources encompass a wide range of topics, including pathways, protein function, interacting proteins, related articles, and research areas. These invaluable resources serve as a knowledge hub, empowering researchers to deepen their understanding of IL-12 family receptors and their crucial roles in physiological processes.
Our Featured Products
| Cat.# | Product name | Species | Source (Host) | Tag |
|---|---|---|---|---|
| IL12RB1-3230H | Active Recombinant Human IL12RB1 protein, His-tagged | Human | HEK293 | His |
| IL12RB2-73H | Recombinant Human IL12RB2 protein, Fc-tagged | Human | HEK293 | Fc |
| Il12rb2-4006M | Active Recombinant Mouse Il12rb2 protein, His-tagged | Mouse | HEK293 | His |
| IL23R-3082H | Recombinant Human IL23R protein, His-tagged | Human | E.coli | His |
| IL23R-8493C | Active Recombinant Cynomolgus/Rhesus IL23R protein, hFc-tagged | Cynomolgus/Rhesus | HEK293 | hFc |
| IL6ST-787H | Recombinant Human IL6ST protein, His-tagged | Human | E.coli | His |
| Il6st-4091R | Recombinant Rat Il6st, Fc-His tagged | Rat | Human Cell | Fc/His |
About IL-12 Family Receptors
The IL-12 family of cytokines, including IL-12, IL-23, IL-27, and IL-35, exert their biological effects by binding to specific receptors on target cells. Here's an introduction to the IL-12 family receptors:
IL-12 Receptor β1 (IL-12Rβ1)
IL-12Rβ1 is a common subunit shared by the receptors of IL-12 and IL-23. It is a transmembrane protein expressed on various immune cells, including T cells, NK cells, and antigen-presenting cells (APCs). IL-12Rβ1 forms a heterodimeric receptor with IL-12Rβ2 for IL-12 signaling and with IL-23R for IL-23 signaling. Upon ligand binding, IL-12Rβ1 initiates intracellular signaling pathways, including the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, leading to the activation of downstream signaling cascades.
IL-12 Receptor β2 (IL-12Rβ2)
IL-12Rβ2 is a specific subunit that pairs with IL-12Rβ1 to form the functional receptor for IL-12 signaling. It is primarily expressed on T cells and NK cells. The binding of IL-12 to the IL-12 receptor complex (IL-12Rβ1/IL-12Rβ2) triggers signaling events that promote T cell differentiation into the Th1 subset and enhance the cytotoxic activity of NK cells.
IL-23 Receptor (IL-23R)
IL-23R is a specific subunit that pairs with IL-12Rβ1 to form the functional receptor for IL-23 signaling. It is predominantly expressed on T cells, particularly Th17 cells. Binding of IL-23 to the IL-23 receptor complex (IL-12Rβ1/IL-23R) activates downstream signaling pathways, including the JAK/STAT pathway, which leads to the expansion, survival, and activation of Th17 cells. IL-23R signaling is crucial for the pathogenesis of autoimmune and inflammatory diseases associated with Th17 responses.
IL-27 Receptor α (IL-27Rα)
IL-27Rα is the specific receptor for IL-27 signaling. It is expressed on various immune cells, including T cells, B cells, NK cells, and APCs. IL-27Rα forms a heterodimeric receptor complex with gp130, a shared signaling subunit among several cytokine receptors. Binding of IL-27 to IL-27Rα/gp130 initiates intracellular signaling, modulating immune responses and inflammation. IL-27Rα signaling influences the balance between Th1 and Th17 responses and promotes the differentiation and function of regulatory T cells (Tregs).
Glycoprotein 130 (gp130)
gp130 (alternatively known as IL6ST, CD130, CDW130, GP130, IL-6RB.) is a shared signaling subunit that is involved in the signaling of multiple cytokines, including IL-6, IL-11, IL-27, and IL-35. It forms a heterodimeric receptor complex with specific receptor subunits, such as IL-27Rα for IL-27 signaling. gp130 is widely expressed on various cell types, including immune cells and non-immune cells. Activation of gp130-associated receptors triggers intracellular signaling pathways, including the JAK/STAT pathway, to mediate the biological effects of respective cytokines.
Understanding the receptors of IL-12 family cytokines is essential for deciphering their signaling mechanisms and the regulation of immune responses. The activation of these receptors by their specific ligands initiates signaling cascades that influence immune cell differentiation, inflammation, and the maintenance of immune homeostasis.
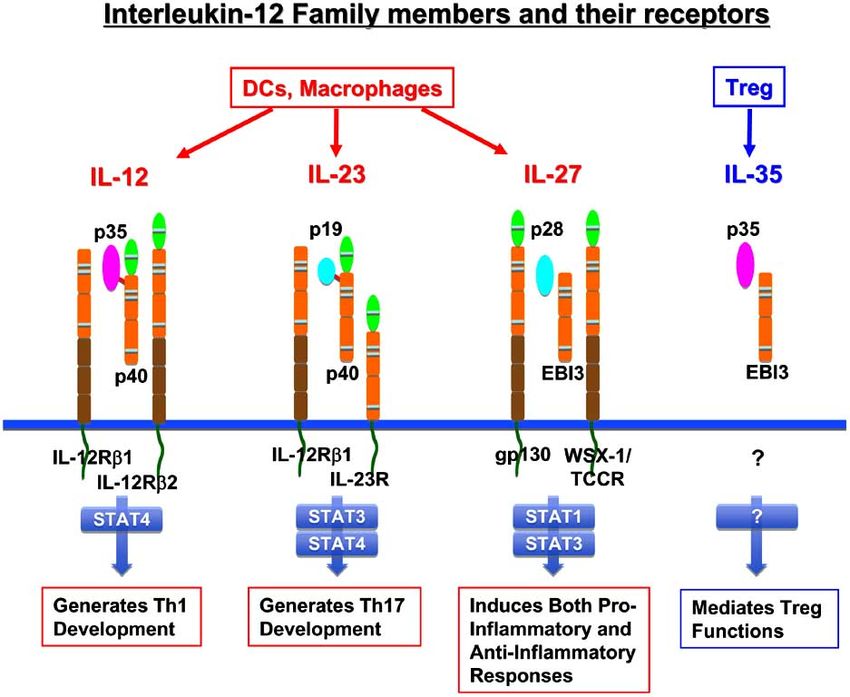 Fig.1 Main characteristics of members of the IL-12 family. (Nagai H, et al., 2010)
Fig.1 Main characteristics of members of the IL-12 family. (Nagai H, et al., 2010)
Signaling Pathways and Mechanisms of Action of IL-12 Family Receptors
IL-12 Signaling Pathway
IL-12 binds to the IL-12 receptor complex, consisting of IL-12Rβ1 and IL-12Rβ2 subunits. This binding activates Janus kinase 2 (JAK2) and Tyrosine kinase 2 (Tyk2), which phosphorylate specific tyrosine residues on the receptor subunits. Phosphorylated receptor subunits create docking sites for signal transducer and activator of transcription 4 (STAT4). STAT4 is recruited, phosphorylated by JAKs, and forms a dimer that translocates into the nucleus. In the nucleus, STAT4 binds to specific DNA sequences, leading to the transcription of target genes, such as IFN-γ. The activation of the IL-12 signaling pathway promotes Th1 cell differentiation and enhances the cytotoxic activity of NK cells.
IL-23 Signaling Pathway
IL-23 binds to the IL-23 receptor complex, composed of IL-23R and IL-12Rβ1 subunits. This binding activates JAK2 and Tyk2, which phosphorylate tyrosine residues on the receptor subunits. Phosphorylated receptor subunits create docking sites for various signaling molecules, including STAT3, JAK2, and Tyk2. STAT3 is a key mediator of IL-23 signaling and is phosphorylated by JAK2 and Tyk2. Phosphorylated STAT3 forms a dimer and translocates into the nucleus, where it binds to specific DNA sequences, regulating the transcription of target genes involved in Th17 cell differentiation and activation.
IL-27 Signaling Pathway
IL-27 binds to the IL-27 receptor complex, which consists of IL-27Rα and gp130 subunits. This binding activates JAK1 and Tyk2, leading to the phosphorylation of receptor subunits. Phosphorylated receptor subunits create docking sites for STAT1 and STAT3. Both STAT1 and STAT3 are recruited, phosphorylated by JAKs, and form dimers that translocate into the nucleus. In the nucleus, these STAT proteins regulate the transcription of target genes, which can modulate immune responses, promote Treg differentiation, and affect the balance between Th1 and Th17 responses.
IL-35 Signaling Pathway
IL-35 signals through the IL-12 receptor complex, composed of IL-12Rβ2 and IL-27Rα subunits. The specific signaling mechanisms activated by IL-35 are not fully understood. However, it is thought that IL-35 can utilize the JAK/STAT pathway, similar to IL-12 and IL-27.
It's important to note that IL-12 family cytokines can activate other signaling pathways and molecular cascades in addition to the ones mentioned above, depending on the cell type and context. These include MAPK, PI3K/Akt, and NF-κB pathways, which can further modulate cellular responses and gene expression.
Understanding the signaling pathways and mechanisms of IL-12 family receptors provides insights into the regulation of immune responses and the development of targeted therapies for immune-related disorders.
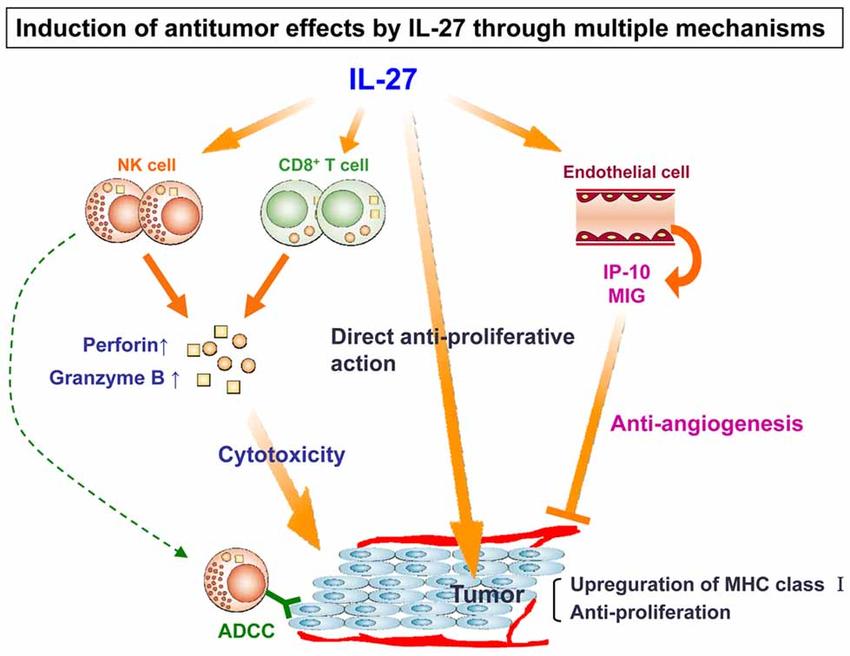 Fig.2 Possible mechanisms of the antitumor effects of IL-27 on melanoma cells. (Nagai H, et al., 2010)
Fig.2 Possible mechanisms of the antitumor effects of IL-27 on melanoma cells. (Nagai H, et al., 2010)
If you have any questions, requirements, or cooperation intentions, please feel free to contact us. We very much look forward to working with you and helping you achieve research and commercial success.
Related References:
- Nagai H, Oniki S, Fujiwara S, et al. Antitumor activities of interleukin-27 on melanoma. Endocr Metab Immune Disord Drug Targets. 2010;10(1):41-46. doi:10.2174/187153010790827920
- Gee K, Guzzo C, Che Mat NF, Ma W, Kumar A. The IL-12 family of cytokines in infection, inflammation and autoimmune disorders. Inflamm Allergy Drug Targets. 2009;8(1):40-52. doi:10.2174/187152809787582507