


BCL2 Family Classification
Apoptosis is a tightly regulated process that allows the elimination of damaged, infected, or unnecessary cells without triggering an inflammatory response. The BCL2 family of proteins is central to the intrinsic (mitochondrial) pathway of apoptosis and controls mitochondrial outer membrane permeabilization (MOMP), a critical event leading to cell death. Members of this family share conserved regions known as BCL2 homology (BH) domains that mediate their interactions and functions.
BCL2 family proteins are categorized based on their structural features and functional roles in apoptosis:
- Anti-apoptotic Proteins: These proteins contain four BCL2 homology (BH) domains (BH1-BH4) and function to inhibit apoptosis by preventing mitochondrial outer membrane permeabilization (MOMP). Key members include BCL2, BCL-xL, and MCL1.
- Pro-apoptotic Effector Proteins: Also known as multi-domain pro-apoptotic proteins, they contain BH1, BH2, and BH3 domains and promote apoptosis by inducing MOMP. The major members are BAX and BAK.
- Pro-apoptotic BH3-only Proteins: Act as initiators of apoptosis by sensing cellular stress and activating effector proteins or inhibiting anti-apoptotic proteins. Members such as BAD, BID, BIM, PUMA, NOXA, BIK, BMF, and HRK contain only the BH3 domain, which is crucial for their interaction with other BCL2 family proteins.
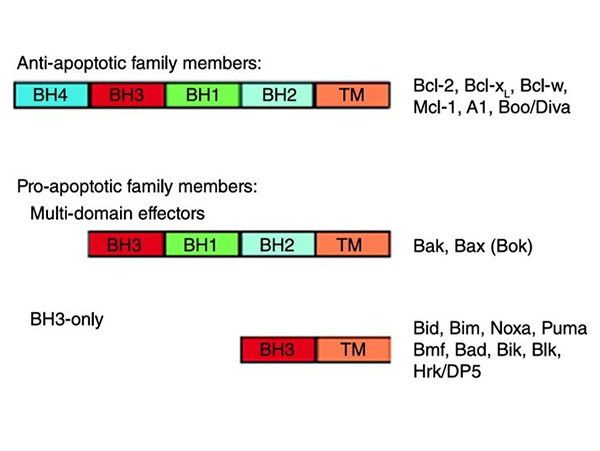
Figure 1. The Bcl-2 protein family is classified into anti-apoptotic family members, which contain four BH homology domains, and pro-apoptotic family members, which contain either three BH homology domains (‘multi-domain effectors’) or only the third BH domain (‘BH3-only’). Illustrated are cartoon schematic diagrams of the different groups, and a list of the members of each group. Most members have a C-terminal hydrophobic domain (TM) which aids association with the mitochondrial membrane. Exceptions are A1 and many of the BH3-only proteins (Bid, Bad, Bmf, NOXA and PUMA). Note: The Bcl-2 protein Bok is a member of the multidomain Bcl-2 protein family; however, there is little evidence to suggest that Bok functions in a manner similar to Bak and Bax (Parsons and Green, 2010)
Anti-apoptotic Proteins
The anti-apoptotic proteins BCL2, BCL-xL and MCL1 contain all four BH domains. The three-dimensional structure of BCL-xL shows that the protein consists of two central hydrophobic α-helices (α5 and α6) surrounded by five amphipathic helices (α1, α2, α3, α4, and α7) and arranged so that the BH1, BH2, and BH3 regions form a hydrophobic pocket to which a BH3 domain can bind. It should be noted that the NMR structure of BCL-xL complexed with a peptide derived from the BH3 domain of the pro-apoptotic protein BAX was obtained in solution. Considering that anti-apoptotic proteins have a hydrophobic C-terminal transmembrane domain that targets them to intracellular membranes, the binding conformation may differ between a solution structure and a membrane-bound structure.
The BH4 domain of BCL-xL forms an amphipathic helix on the opposite side of the molecule and may further stabilize the hydrophobic pocket. Other anti-apoptotic members with similar homology domains are predicted to have a similar tertiary structure. Between the BH3 and BH4 domains is an unstructured, flexible loop of approximately 60 residues in length. This region is a site of negative regulation, but is not essential for the anti-apoptotic function of at least BCL-xL. Although the mechanism by which such regulation occurs is still unclear, phosphorylation of sites within the loop by serine/threonine kinases may be one way to modulate anti-apoptotic function. Alternatively, anti-apoptotic proteins may be converted to death promoters as a consequence of loss of the BH4 domain by cleavage of putative caspase sites within the loop region. Loss of the BH4 domain in BCL2, for example, reduces or abolishes its anti-apoptotic function and may confer death-promoting properties.
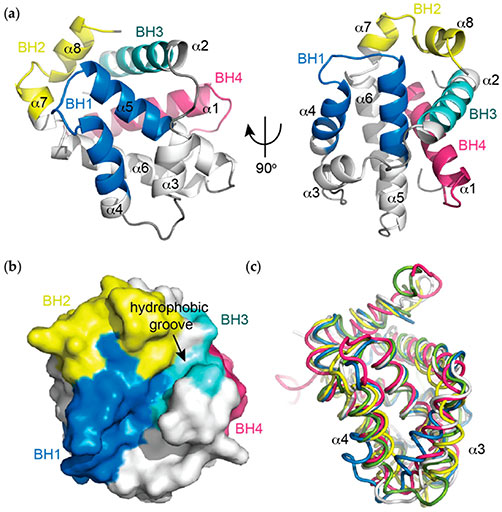
Figure 2. (a) Helical representation of apo BCL-xL (PDB ID: 1MAZ). The BCL-2 homology (BH) domains (coloured), make significant contributions to defining the tertiary structure of BCL-2 pro-survival proteins. The α5 and α6 helices form a central hairpin surrounded on either side by the other helices; (b) Surface representation of apo BCL-xL demonstrating the canonical hydrophobic binding groove created mainly by helices α3 and α4 with α5 forming the base, which is critical for mediating interactions with the pro-apoptotic proteins of the BCL-2 family; (c) Overlay of apo structures of the pro-survival members of the BCL-2 family demonstrating idiosyncrasies in the orientations of the α3 and α4 helices that line the hydrophobic groove. In particular, notable differences in the orientation of α3 and α4 are observed. BCL-xL (PDB ID: 1MAZ, blue); BCL-2 (PDB ID: 1GJH, white); BCL-w (PDB ID: 1O0L, green); MCL-1 (PDB ID: 1WSX, pink); BFL-1 (PDB ID: 5WHI, yellow). (Lee and Fairlie, 2019)
The anti-apoptotic proteins BCL2 and BCL-xL are similar in structure and size (~25 KDa), with long protein half-lives (more than 20 hours). MCL1 is a larger protein (37 KDa) with a short half-life (~30 minutes) that is regulated by the proteasome-dependent pathway. MCL1 is poly-ubiquitinated by the MCL1 ubiquitin ligase E3 (MULE) and subsequently targeted for proteasomal degradation. Interestingly, MULE contains a BH3 domain that allows it to selectively target MCL1 but not other anti-apoptotic proteins.
Phosphorylation of MCL1 may regulate its proteasomal degradation. For example, GSK-3-mediated phosphorylation of MCL1 induces the ubiquitination and degradation of MCL1 and may affect the recognition of MCL1 by MULE. Phosphorylation of MCL1 by ERK and JNK is thought to occur upstream of GSK-3 and act as an additional regulatory level of MCL1 stabilization. ERK phosphorylates MCL1 at threonine 163 to slow the turnover rate of MCL1, thereby prolonging its protective effects. In contrast, phosphorylation of MCL1 at serine-121 by JNK inactivates MCL1. These data suggest that phosphorylation of MCL1 plays an important role in its half-life and antiapoptotic activity.
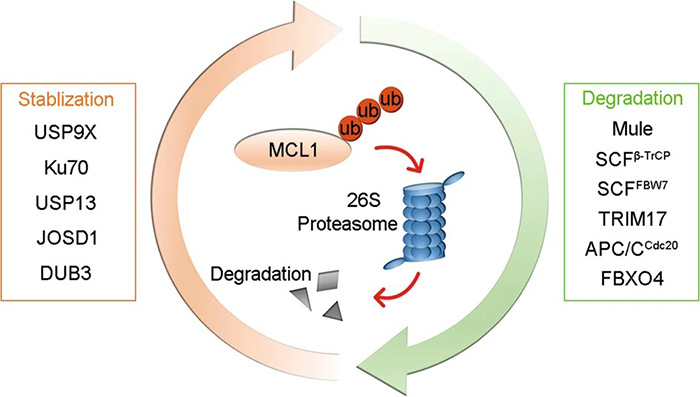
Figure 3. E3 ligases (Mule, SCFβ-TrCP, SCFFBW7, TRIM17, APC/CCdc20, and FBXO4) promote the ubiquitination and degradation of MCL1, whereas deubiquitinases (USP9X, Ku70, USP13, JOSD1, and DUB3) deubiquitinate MCL1 to prevent its degradation. (Wu et al., 2020)
Pro-apoptotic Effector Proteins: BAX and BAK
BAX and BAK are the two multi-domain pro-apoptotic proteins responsible for permeabilizing the outer mitochondrial membrane. They are largely redundant in function, as loss of either BAX or BAK has little effect in most cells and tissues. However, the absence of both proteins blocks apoptosis in many cell types, suggesting the importance of BAX and BAK in the intrinsic apoptotic pathway.
In viable cells, BAX and BAK are present as monomers. Inactive BAX resides in the cytosol or is loosely associated with the membrane with its C-terminal tail folded back into the hydrophobic BAX groove formed by the BH1, BH2, and BH3 domains. Upon apoptotic stimulation, BAX undergoes a conformational change that releases the C-terminal tail, allowing to insert into the mitochondrial outer membrane. In contrast to BAX, BAK monomers are mitochondria-resident proteins. BAK also undergoes allosteric conformational activation in response to death signals.
Once activated, both BAX and BAK homo-oligomerize to form pores in the mitochondrial outer membrane. Through these pores, cytochrome c is released into the cytosol, ultimately leading to the activation of the caspase cascade.
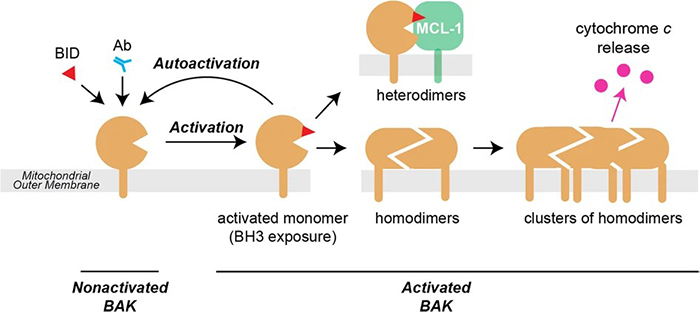
Figure 4. In healthy cells, nonactivated BAK is inserted into the mitochondrial outer membrane via its α9 transmembrane domain. Activation can be triggered by BH3-only proteins such as BID (by binding to the α2–α5 groove) or by antibody (binding to the N-terminus of the α1–α2 loop). The major conformation changes that occur during activation include separation of three components—the N-terminal α1–helix, the α2–α5 core, and the α6–α9 latch—leaving the protein with an exposed BH3 domain (α2; red triangle) and a modified α2–α5 hydrophobic groove. Once activated, the BAK monomer can bind to other family members to form either homodimers that promote apoptosis, or heterodimers with a pro-survival relative (here MCL-1) that do not contribute to pore formation. The activated monomer may also activate (autoactivate) additional BAK (or BAX) molecules. (Iyer et al., 2020)
BH3-Only Proteins
BH3-only proteins are key pro-apoptotic members of the BCL2 family, acting as sentinels that sense and respond to various cellular stress signals to induce apoptosis. Their defining feature is the BH3 domain, a short α-helical sequence that is essential for their interaction with other BCL-2 family proteins. This domain enables BH3-only proteins to bind anti-apoptotic counterparts such as BCL2 and BCL-xL, neutralizing their function and promoting the activation of pro-apoptotic effectors such as BAX and BAK, leading to mitochondrial outer membrane permeabilization (MOMP) and subsequent cell death.
The regulation of BH3-only proteins is multifaceted and includes transcriptional upregulation, post-translational modifications, and controlled subcellular localization. For example, growth factor deprivation in neurons can induce the expression of HRK and BIM through a c-Jun N-terminal kinase (JNK)-dependent pathway. Similarly, the transcription factor FOXO3A regulates the expression of BIM and NOXA by integrating signals from different stress stimuli.
Post-translational modifications further modulate the activity of BH3-only proteins. For example, BAD is phosphorylated by kinases such as AKT, PKA, and JNK. When phosphorylated, BAD associates with 14-3-3 proteins in the cytosol, preventing its interaction with mitochondrial targets and thus inhibiting its pro-apoptotic function. Conversely, dephosphorylation releases BAD, allowing it to translocate to the mitochondria and promote apoptosis.
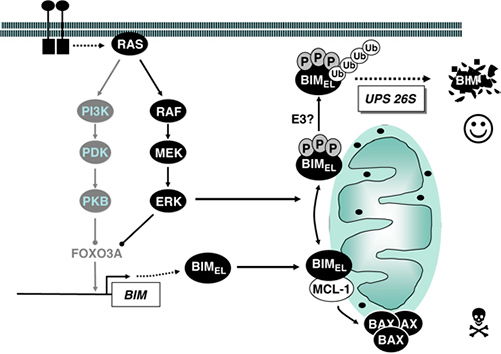
Figure 5. Regulation of BIM binding properties and stability by ERK1/2. The pro-apoptotic BH3-only protein BIM is expressed de novo following cytokine withdrawal and binds to pro-survival proteins such as MCL-1, thereby releasing BAX or BAK to promote cell death. ERK1/2-catalysed phosphorylation of FOXO3A promotes its proteasomal degradation, thereby preventing FOXO3A-dependent expression of BIM. In addition, BIMEL, the most abundant BIM splice variant, is phosphorylated directly by ERK1/2 on at least three different sites. This promotes the dissociation of BIMEL from pro-survival proteins and targets BIMEL for poly-ubiquitination through the 26S proteasome. (Balmanno and Cook, 2009)
The subcellular localization of BH3-only proteins is closely linked to their activation status. BIM isoforms, such as BIMEL and BIML, are typically sequestered at the microtubule-associated dynein motor complex via binding to the dynein light chain LC8. Upon apoptotic stimuli, these isoforms are released and translocate to the mitochondria where they exert their pro-apoptotic effects.
Recent research has expanded our understanding of BH3-only proteins. Studies have shown that certain BH3-only proteins, such as PUMA and NOXA, play a critical role in DNA damage-induced apoptosis, acting downstream of p53 activation. In addition, the discovery of novel BH3-only proteins, such as BNIP5, and their unique interactions with other BCL2 family members continue to refine our understanding of apoptotic regulation (Rühl et al., 2025).
The specificity and diversity of BH3-only proteins underscore their role as specialized sentinels for various apoptotic signals, including DNA damage, growth factor deprivation, and cytoskeletal perturbations. Their precise regulation and targeted activation make them critical components in maintaining cellular homeostasis and responding to pathological conditions.
Interactions and Regulatory Mechanisms
The interactions between these proteins are finely tuned to determine cell fate. Under normal conditions, anti-apoptotic proteins bind and sequester pro-apoptotic members, preventing unwarranted cell death. Upon apoptotic stimuli, BH3-only proteins are upregulated or activated; they can directly activate BAX and BAK or neutralize anti-apoptotic proteins, thereby promoting MOMP and subsequent apoptosis.
The regulatory mechanisms governing these interactions include transcriptional control, post-translational modifications (such as phosphorylation and ubiquitination), and subcellular localization. For example, phosphorylation of BAD by kinases such as AKT leads to its sequestration by 14-3-3 proteins, preventing it from inhibiting BCL2 and thus promoting cell survival.
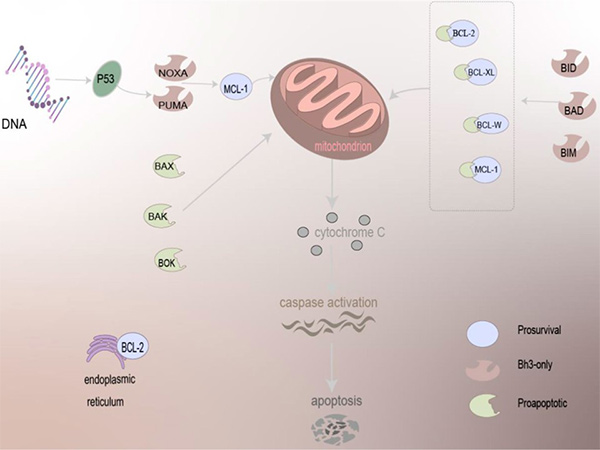
Figure 6. The mechanism of BCL-2 family proteins in regulating apoptosis. Driven by BCL-2 family proteins, which are primarily localized to mitochondria and present on the ER, pro-apoptotic family proteins can act directly on the mitochondria to trigger the apoptosis mechanism, and anti-apoptotic proteins need to work together with other proteins to cause apoptosis, release cytochrome C, and activation of caspase of apoptosis. (Qian et al., 2022)
References
- Antignani A, Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Current Opinion in Cell Biology. 2006;18(6):685-689. doi:10.1016/j.ceb.2006.10.004
- Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16(3):368-377. doi:10.1038/cdd.2008.148
- Cheng E H Y, Kirsch D G, Clem R J, et al. Conversion of Bcl-2 to a Bax-like death effector by caspases[J]. Science, 1997, 278(5345): 1966-1968.
- Cheng EHY, Kirsch DG, Clem RJ, et al. Conversion of bcl-2 to a bax-like death effector by caspases. Science. 1997;278(5345):1966-1968. doi:10.1126/science.278.5345.1966
- Iyer S, Uren RT, Dengler MA, et al. Robust autoactivation for apoptosis by BAK but not BAX highlights BAK as an important therapeutic target. Cell Death Dis. 2020;11(4):1-13. doi:10.1038/s41419-020-2463-7
- Lee EF, Fairlie WD. The structural biology of bcl-xl. IJMS. 2019;20(9):2234. doi:10.3390/ijms20092234
- McDonnell T J, Deane N, Platt F M, et al. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation[J]. Cell, 1989, 57(1): 79-88.
- McDonnell TJ, Deane N, Platt FM, et al. Bcl-2-immunoglobulin transgenic mice demonstrate extended b cell survival and follicular lymphoproliferation. Cell. 1989;57(1):79-88. doi:10.1016/0092-8674(89)90174-8
- Parsons MJ, Green DR. Mitochondria in cell death. Brown GC, Murphy MP, eds. Essays in Biochemistry. 2010;47:99-114. doi:10.1042/bse0470099
- Pistritto G, Trisciuoglio D, Ceci C, Garufi A, D’Orazi G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging. 2016;8(4):603-619. doi:10.18632/aging.100934
- Puthalakath H, Villunger A, O’Reilly LA, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293(5536):1829-1832. doi:10.1126/science.1062257
- Qian S, Wei Z, Yang W, Huang J, Yang Y, Wang J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front Oncol. 2022;12:985363. doi:10.3389/fonc.2022.985363
- Wu X, Luo Q, Liu Z. Ubiquitination and deubiquitination of MCL1 in cancer: deciphering chemoresistance mechanisms and providing potential therapeutic options. Cell Death Dis. 2020;11(7):1-11. doi:10.1038/s41419-020-02760-y
Contact us or send an email at for project quotations and more detailed information.
Quick Links
-

Papers’ PMID to Obtain Coupon
Submit Now -

Refer Friends & New Lab Start-up Promotions