
Neuronal Lineage Markers
Related Symbol Search List
- L1CAM
- NEUROD1
- ACE
- ALCAM
- DLG4
- GAD1
- Gad2
- GRIN1
- HOXA1
- INA
- LHX5
- Latexin
- MAP2
- MAPT
- MSX1
- NEFH
- NEFL
- NEFM
- NELL1
- NELL2
- NEUROD2
- NRG2
- SNCA
- STX1A
- SYP
- Tyrosine Hydroxylase
- Thy1
- TLE3
- TUBB3
- VAMP1
- VAMP2
- VAMP5
- VAMP7
- VAMP8
Immunology Background
Background
Neuronal Lineages
Neuronal lineages refer to the developmental paths that neural stem cells and progenitor cells follow as they differentiate into various types of neurons in the central nervous system. These lineages are crucial in shaping the complex network of neurons that govern cognitive and motor functions in the human body.
Key Points of Neuronal Lineages
1. Neural Stem Cells (NSCs)
Neural stem cells are the source of all neurons in the brain. They have the ability to self-renew and differentiate into various neuronal lineages.
2. Progenitor Cells
Progenitor cells are intermediate cells derived from neural stem cells. They are more restricted in their differentiation potential compared to stem cells.
3. Types of Neurons
Neuronal lineages give rise to different types of neurons, such as sensory neurons, motor neurons, interneurons, and projection neurons, each with unique functions and characteristics.
4. Developmental Process
During development, neural stem cells undergo asymmetric division to generate progenitor cells that further differentiate into specific neuronal lineages based on intrinsic genetic programs and extrinsic signals from the environment.
5. Regional Specification
Neural progenitor cells in different regions of the brain give rise to distinct neuronal lineages. For example, cortical neural progenitors produce neurons that populate the cerebral cortex, while those in the spinal cord generate spinal cord neurons.
6. Functional Diversity
Neuronal lineages contribute to the functional diversity of the nervous system by forming intricate circuits and networks that underlie sensory perception, motor control, cognition, and behavior.
7. Regulation and Maintenance
The regulation and maintenance of neuronal lineages are influenced by a complex interplay of transcription factors, signaling molecules, and epigenetic modifications that control cell fate decisions and neuronal maturation.
Understanding neuronal lineages is essential for unraveling the cellular basis of brain function, development of therapeutic strategies for neurological disorders, and advancements in regenerative medicine. By dissecting the intricate process by which neural stem cells give rise to diverse neuronal types, researchers can gain insights into the fundamental principles governing brain development and function.
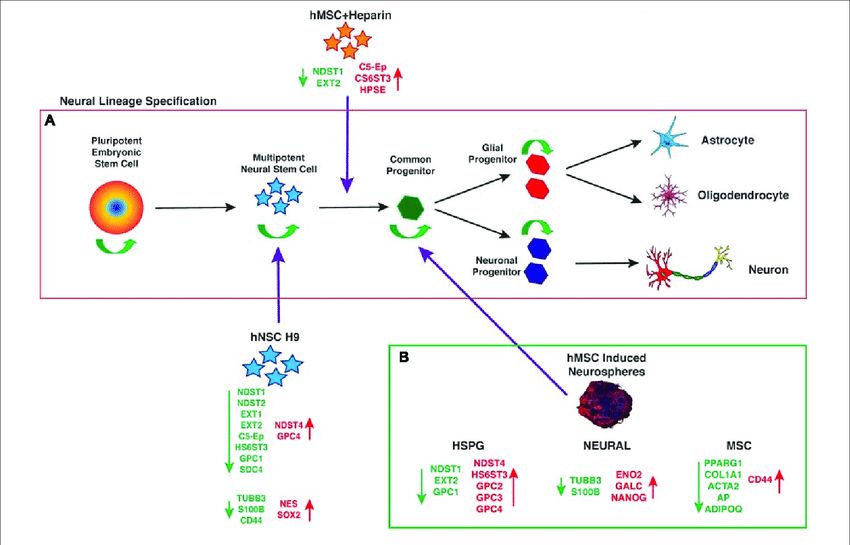 Fig.1 Neural lineage specification. (Okolicsanyi RK, et al., 2018)
Fig.1 Neural lineage specification. (Okolicsanyi RK, et al., 2018)Neuronal Lineage Markers
Neuronal lineage markers are specific proteins or genes that are expressed at different stages of neuronal development, helping to identify and track the differentiation of neural stem cells and progenitor cells into mature neurons. These markers play a crucial role in understanding the complex process of neuronal lineage specification and maturation in the central nervous system.
Key Points of Neuronal Lineage Markers
1. Identification and Characterization
Neuronal lineage markers serve as molecular signatures that allow researchers to identify and characterize different stages of neuronal development, from neural stem cells to mature neurons.
2. Cell Fate Determination
These markers help in determining the fate of neural progenitor cells by indicating the specific neuronal lineage they are destined to differentiate into, such as excitatory neurons, inhibitory neurons, or glial cells.
3. Developmental Stage Specificity
Neuronal lineage markers are often expressed in a stage-specific manner, providing insights into the timing and progression of neuronal differentiation during embryonic development and in adult neurogenesis.
4. Functional Diversity
Different neuronal lineage markers are associated with distinct neuronal subtypes with specialized functions and connectivity patterns, contributing to the complexity and diversity of the nervous system.
5. Research and Clinical Applications
Studying neuronal lineage markers is essential for basic neuroscience research, as well as for potential applications in regenerative medicine, drug discovery, and understanding the pathophysiology of neurological disorders.
6. Examples of Neuronal Lineage Markers
- Nestin: A marker of neural stem cells and early progenitor cells.
- Doublecortin (DCX): Expressed in migrating neuroblasts and immature neurons.
- NeuN (RBFOX3): A marker for mature neurons in the central nervous system.
- Olig2: Marks oligodendrocyte progenitor cells involved in myelination.
- GFAP: A marker for astrocytes, a type of glial cell in the brain.
Understanding the expression patterns and functions of neuronal lineage markers is essential for dissecting the complex process of neuronal development, from cell fate determination to the formation of functional neuronal circuits. By unraveling the role of these markers, researchers can gain valuable insights into neurogenesis, brain plasticity, and the underlying mechanisms of neurological diseases.
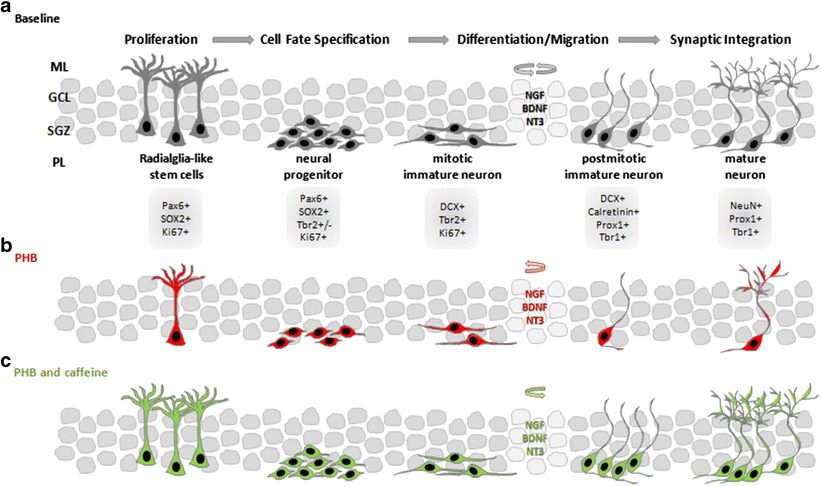 Fig.2 The cellular developmental stages of the neural lineage revolve around a network of neuronal mediators and transcription factors. (Endesfelder S, et al., 2018)
Fig.2 The cellular developmental stages of the neural lineage revolve around a network of neuronal mediators and transcription factors. (Endesfelder S, et al., 2018)Neural lineage cell development a passes through various phases of proliferation, cell fate specification, and differentiation which are associated with migration, and synaptic integration, orchestrated about a network of neuronal mediators and transcription factors, which are expressed dependent on maturation stage. Specific transcription factors for hippocampal neurogenesis are Pax6, SOX2, Tbr1/2, and Prox1, expressed in conjunction with the neuronal marker DCX for mitotic immature neurons, calretinin for postmitotic immature neurons, and NeuN for mature neurons.
Common Neuronal Lineage Markers
Here are some common markers used to characterize neural lineage:
Different Molecular Types
| Type | Details |
|---|---|
| Transcription Factors |
|
| Cell Adhesion Molecules |
|
| Neuronal Markers |
|
| Neurotransmitter Synthesizers |
|
| Synaptic Proteins |
|
| Myelin and Axonal Protection |
|
| Extracellular Matrix and Cell Signaling |
|
| Developmental and Plasticity Markers |
|
| Cell Surface Markers |
|
| Functional Proteins |
|
Different Stages of Neuronal Development
Here is a comprehensive list of markers commonly associated with different types of neuronal lineages, including neural progenitor cells, early differentiated neurons, intermediate differentiated neurons, and mature neurons. These markers serve as molecular signatures that help identify and characterize various stages of neuronal development in the central nervous system:
| Stage | Details |
|---|---|
| Neural Progenitor Cells |
|
| Early Differentiated Neurons |
|
| Intermediate Differentiated Neurons |
|
| Mature Neurons |
|
| General Neuronal Markers (expressed across various stages) |
|
This list provides a broad overview of markers associated with different stages of neuronal development, from neural progenitor cells to mature neurons, aiding in the characterization and understanding of diverse neuronal lineages in the central nervous system.
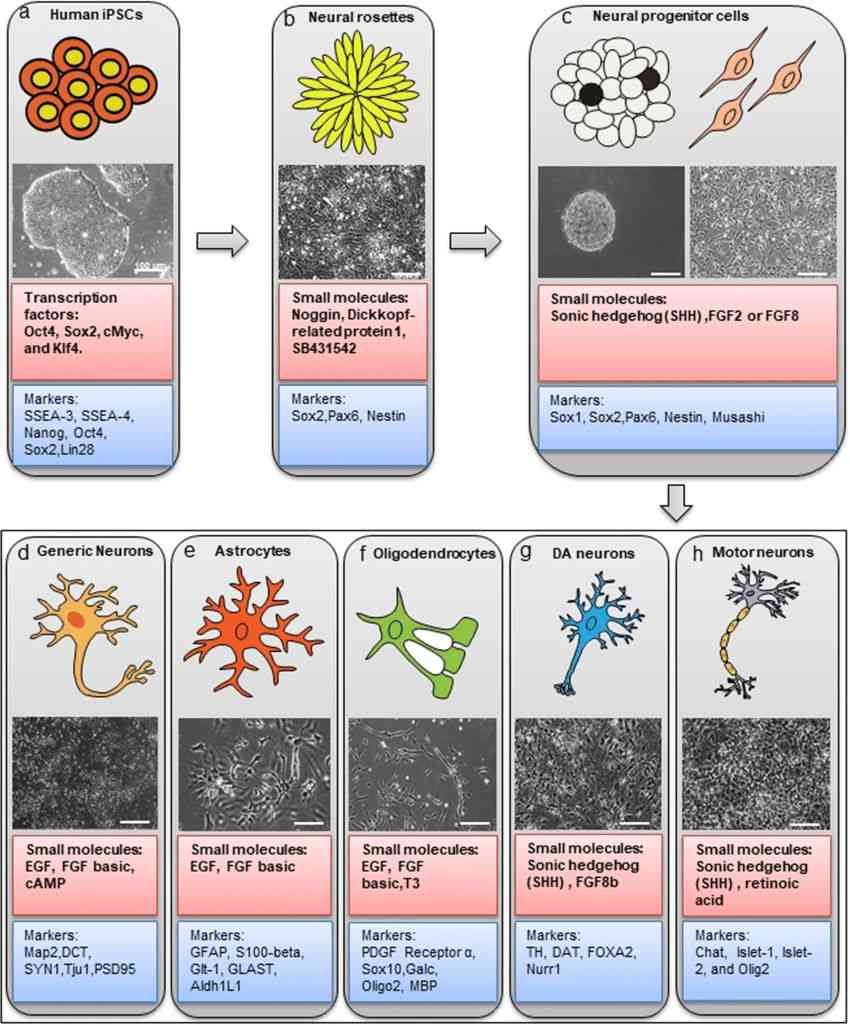 Fig.3 Sequence of cell types of neuronal lineage and lineage-specific markers. (Liang X, et al., 2020)
Fig.3 Sequence of cell types of neuronal lineage and lineage-specific markers. (Liang X, et al., 2020)Applications of Neuronal Lineage Markers
Neuronal lineage markers are crucial tools in neuroscience for tracing the origins and developmental pathways of neurons. Their applications span various domains, including research, medicine, and regenerative therapies. Here are some key applications:
1. Developmental Biology
Tracing Neuronal Development: Lineage markers help researchers track the differentiation of neural stem cells (NSCs) into specific neuronal types during development, elucidating the mechanisms that drive neurogenesis.
Understanding Neuronal Diversity: By marking different progenitor populations, scientists can study the generation of diverse neuron types and their functional implications in various brain regions.
2. Neural Circuit Mapping
Identifying Cell Connections: Neuronal lineage markers can be used to trace neuronal pathways and elucidate the connectivity between different neuronal populations, enhancing our understanding of circuit function and organization.
3. Disease Modeling
Understanding Neurodevelopmental Disorders: Markers allow researchers to study how abnormal lineage specification contributes to disorders such as autism, schizophrenia, or epilepsy.
Tracking Disease Progression: In neurodegenerative diseases, lineage markers can help assess how specific neuronal populations degenerate over time.
4. Stem Cell Research
Identifying NSCs: Lineage markers can distinguish between NSCs and more differentiated progenitors, aiding in the selection of cells for transplantation or therapeutic purposes.
Optimizing Differentiation Protocols: Researchers can use markers to improve protocols for inducing stem cells to differentiate into specific neuronal types for research or therapeutic applications.
5. Regenerative Medicine
Cell Replacement Therapies: Markers can aid in selecting and characterizing stem or progenitor cells for transplant in treating neurological injuries (e.g., spinal cord injuries) or neurodegenerative diseases (e.g., Parkinson's disease).
Assessing Regeneration: In injury contexts, lineage markers can evaluate whether transplanted or endogenous stem cells successfully differentiate into functional neurons.
6. Theranostics
Monitoring Treatment Efficacy: Lineage markers can be incorporated into imaging techniques to monitor the success of treatments aimed at neurodegenerative diseases or brain injuries, allowing real-time assessment of neuronal repair and regeneration.
7. Pharmacological Research
Drug Development: Understanding how drugs influence specific neuronal lineages can inform the development of targeted therapies for neurological conditions.
8. Functional Studies
Investigation of Neuronal Function: By selectively marking neuronal populations, researchers can study the physiological properties and responses of specific neurons to stimuli, shedding light on their roles in behavior and cognition.
Neuronal lineage markers offer powerful tools for advancing our understanding of neurodevelopment, neural circuitry, and pathophysiology. Their applications span fundamental research to therapeutic innovations, providing critical insights that can lead to better diagnostic and treatment options for various neurological conditions.
Impact of Neuronal Lineage Markers on Neural Pathologies
Neuronal lineage markers are integral in various facets of neural development, repair, and disease, offering critical insights into the complex processes that govern the formation, maintenance, and dysfunction of the nervous system. These markers help identify and track different stages of neuronal development, delineate cell types, and provide a molecular understanding of neural pathologies.
| Fields | Details |
|---|---|
| Neuronal Lineage Markers in Neural Development |
Neurogenesis and Cell Fate Determination Neuronal lineage markers play a key role in tracing the differentiation of neural stem cells into specific neuronal lineages during embryonic development and adult neurogenesis. |
|
Regional Specification and Circuit Formation They aid in identifying distinct neuronal subtypes in different brain regions, contributing to the development of complex neural circuits and networks essential for brain function. |
|
|
Cell Maturation and Synaptic Connectivity Neuronal lineage markers help characterize the maturation of neurons and the establishment of synaptic connections, crucial for information processing in the brain. |
|
| Neuronal Lineage Markers in Neural Repair |
Regenerative Medicine and Stem Cell Therapy By serving as identifiers for specific cell populations, neuronal lineage markers assist in directing stem cell differentiation for neural repair and regeneration in conditions such as spinal cord injuries and neurodegenerative diseases. |
|
Enhancement of Neuroplasticity Understanding the expression of neuronal lineage markers can aid in promoting neuroplasticity and facilitating functional recovery following neural injuries or diseases. |
|
| Neuronal Lineage Markers in Neural Disease |
Diagnostic Biomarkers Certain neuronal lineage markers serve as diagnostic indicators for neurological disorders, enabling early detection, disease monitoring, and personalized treatment strategies. |
|
Pathological Mechanisms Altered expression of neuronal lineage markers in disease states provides insights into the underlying cellular and molecular mechanisms contributing to neurodegenerative disorders like Alzheimer's, Parkinson's, and Huntington's disease. |
|
|
Drug Development Targets Neuronal lineage markers offer potential targets for drug development aimed at modulating specific neuronal populations to mitigate disease progression and symptoms. |
Neuronal lineage markers are indispensable tools in deciphering the intricacies of neural development, repair, and disease. Their roles span from guiding fundamental processes in neural maturation to offering potential avenues for therapeutic interventions in neurological disorders. By unraveling the molecular signatures of neuronal lineages, researchers can advance our understanding of the nervous system and pave the way for innovative strategies in neural regeneration and disease treatment.
Case Study
Case 1: Chang HY, Cheng HY, Tsao AN, Liu C, Tsai JW. Multiple functions of KBP in neural development underlie brain anomalies in goldberg-shprintzen syndrome. Front Mol Neurosci. 2019;12:265.
The Kinesin-binding protein (KBP; also known as KIF1BP or KIAA1279) acts as a regulator for a subset of kinesins crucial in neural development. Research has demonstrated that KBP is present in nearly all tissues with cytoplasmic localization. Autosomal recessive mutations in KIAA1279 are linked to Goldberg-Shprintzen syndrome (GOSHS), a rare neurological disorder characterized by microcephaly, polymicrogyria, intellectual disability, axonal neuropathy, thin corpus callosum, and peripheral neuropathy. The majority of mutations associated with GOSHS are homozygous nonsense mutations that lead to KBP loss-of-function. However, the precise mechanisms through which KBP dysfunction triggers these abnormalities remain incompletely understood.
In a study utilizing in utero electroporation (IUE) to introduce KBP short hairpin RNA (shRNA) along with green fluorescent protein (GFP) into neural progenitor cells of embryonic day (E) 14 mice, brain slices were collected at various developmental stages. Through immunostaining of neuronal lineage markers, it was observed that KBP knockdown did not impact the neural differentiation process. Nonetheless, at 4 days post IUE, a significant number of cells congregated in the intermediate zone (IZ). Additionally, by postnatal day (P) 6, approximately one third of the cells, now mature neurons, were abnormally situated in the white matter (WM), while those that had reached Layer II/III of the cortex displayed hindered dendritic outgrowth and axonal projection. Furthermore, the study revealed that KBP knockdown triggered apoptosis during the postnatal period.
The research findings suggest that the absence of functional KBP leads to abnormalities in neuronal migration, morphogenesis, maturation, and survival, potentially contributing to the observed brain phenotypes in individuals affected by GOSHS.
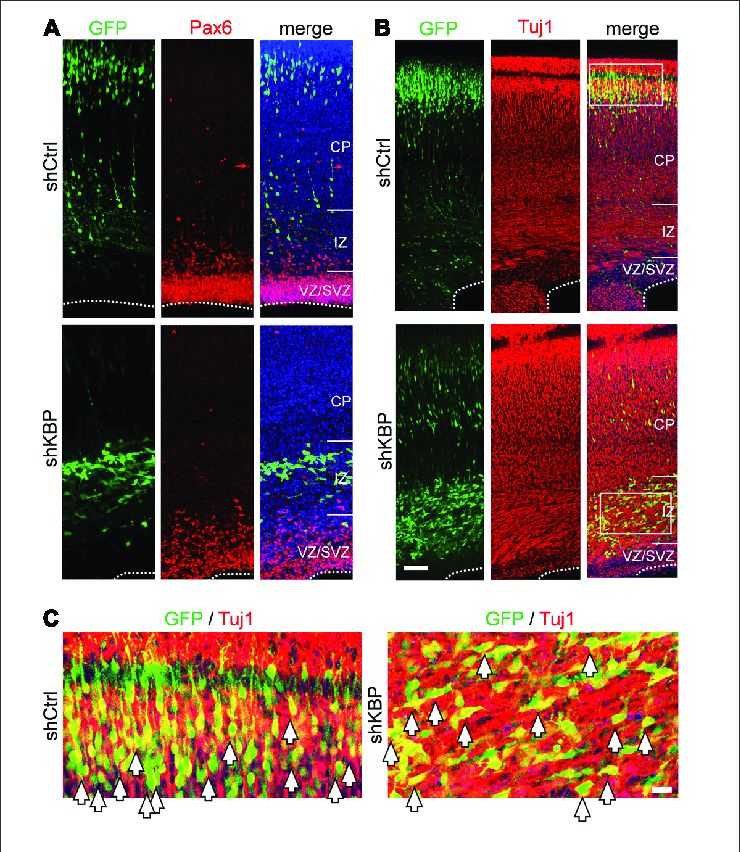 Fig.1 Neuronal lineage of transfected cells after IUE.
Fig.1 Neuronal lineage of transfected cells after IUE.Related References
- Okolicsanyi RK, Oikari LE, Yu C, Griffiths LR, Haupt LM. Heparan sulfate proteoglycans as drivers of neural progenitors derived from human mesenchymal stem cells. Front Mol Neurosci. 2018;11:134.
- Liang X, Kristiansen CK, Vatne GH, Hong Y, Bindoff LA. Patient-specific neural progenitor cells derived from induced pluripotent stem cells offer a promise of good models for mitochondrial disease. Cell Tissue Res. 2020;380(1):15-30.
- Endesfelder S, Weichelt U, Schiller C, Winter K, von Haefen C, Bührer C. Caffeine protects against anticonvulsant-induced impaired neurogenesis in the developing rat brain. Neurotox Res. 2018;34(2):173-187.