
Phospholipases, Small GTPases, and Other Molecules in the Akt Pathway
Related Symbol Search List
- AKT1S1
- APP
- ARRB1
- ARRB2
- CAV1
- CDKN1A
- JAB1
- COPS5
- GSTA4
- HSP27
- HSPB1
- LAX1
- MAPKAP1
- PALLD
- PEBP1
- PLA2G2A
- PLA2G4A
- PLD1
- PRR5
- RAB25
- RAB27A
- GNB2L1
- RALA
- RAP1A
- RAP1B
- RASSF2
- RHEB
- RPTOR
- RRAS2
- SPRY1
- SPRY2
- STIM1
- TOB1
- Vinculin
- WASF1
Immunology Background
About Phospholipases, Small GTPases, and Other Molecules in the Akt Pathway
The Akt pathway, also known as the PI3K/Akt pathway, plays a crucial role in a variety of cellular processes including cell survival, growth, metabolism, and proliferation. While connexins, intracellular kinases, and phosphatases have been extensively studied in the Akt pathway, several other important molecules are involved, such as phospholipases, small GTPases, and others. Exploring the complex network of molecules in the Akt pathway, focusing on phospholipases, small GTPases, and other key components that drive cellular signaling, and understanding the roles and interactions of these molecules is fundamental to unraveling the complex signaling cascades responsible for a wide range of physiological and pathological processes.
Phospholipases
Phospholipases are important regulators of lipid signaling and cellular homeostasis. For example, phospholipase D (PLD) and phospholipase C (PLC) play key roles in mediating signaling events that converge on the Akt pathway. These enzymes regulate the levels of important lipid second messengers that influence cell proliferation, survival, and metabolism.
Small GTPases
Small GTPases, such as members of the Ras, Rho, and Rab protein families, are key signaling molecules that link extracellular stimuli to intracellular responses. These GTPases act as molecular switches, cycling between an active GTP-bound state and an inactive GDP-bound state. In doing so, they regulate various downstream effectors, including components of the Akt pathway. An in-depth study of the complex crosstalk between small GTPases and the Akt pathway can reveal multifaceted regulatory mechanisms and their impact on health and disease.
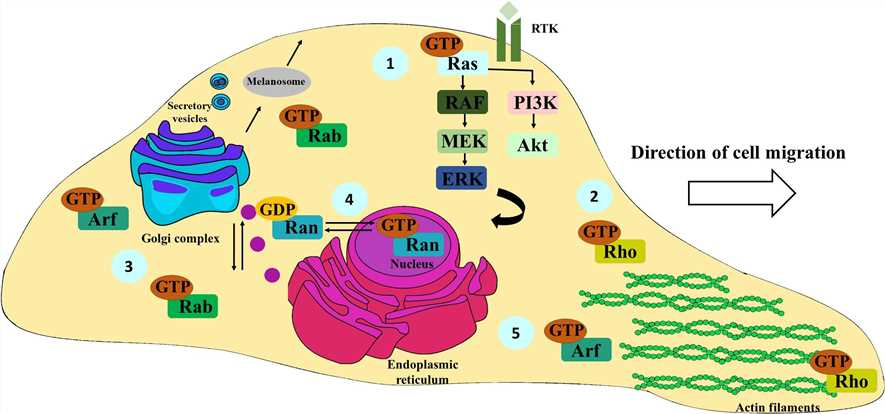 Fig.1 Schematic representation of the most important functions of Ras, Rho, Rab, Ran, and Arf family members. Ras proteins are mainly involved in the activation of the Ras/RAF/MEK/ERK and PI3K-Akt signaling pathways, fundamental for cell survival, proliferation, and differentiation; receptor tyrosine kinase (RTK). (Brito C, et al., 2020)
Fig.1 Schematic representation of the most important functions of Ras, Rho, Rab, Ran, and Arf family members. Ras proteins are mainly involved in the activation of the Ras/RAF/MEK/ERK and PI3K-Akt signaling pathways, fundamental for cell survival, proliferation, and differentiation; receptor tyrosine kinase (RTK). (Brito C, et al., 2020)
Other Key Molecules
In addition to connexins, intracellular kinases, and phosphatases, several other molecules also intricately regulate the Akt pathway, including junction proteins, scaffolding proteins, and molecular chaperones that facilitate the assembly and regulation of signaling complexes. In addition, lipid mediators (e.g., PIP3) and reactive oxygen species (ROS) regulators are critical for Akt pathway activation. Such as AKT1S1 (PRAS40), APP (amyloid precursor protein), PALD (Palladin), PEBP1 (RKIP), RPTOR, RRAS2, SPRY1, STIM1, TOB1, Vinculin, WASF1.
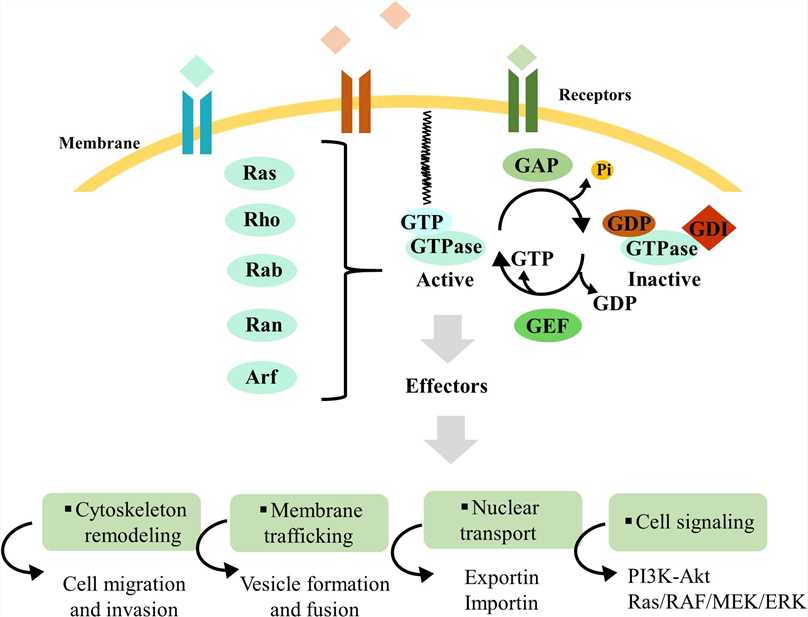 Fig.2 Overview of Ras superfamily small GTPase regulation. Ras superfamily protein regulation mechanisms and downstream interaction with effectors, which control cytoskeleton remodeling, membrane traffic, nuclear transport, and cell signaling. (Brito C, et al., 2020)
Fig.2 Overview of Ras superfamily small GTPase regulation. Ras superfamily protein regulation mechanisms and downstream interaction with effectors, which control cytoskeleton remodeling, membrane traffic, nuclear transport, and cell signaling. (Brito C, et al., 2020)
Available Resources for Phospholipases, Small GTPases, and Other Molecules in the Akt Pathway
Understanding the molecular complexity of the Akt pathway is critical to elucidating its role in cellular processes and disease mechanisms, and Creative BioMart offers a wide range of resources to support researchers studying the Akt pathway and its associated molecules. By utilizing these resources, researchers can deepen their understanding of Akt signaling and potentially develop new therapeutic strategies for a variety of diseases.
Related Products
Creative BioMart offers a wide range of products, including recombinant proteins, cell and tissue lysates, protein pre-coupled magnetic beads, and assay kits to support researchers studying the Akt pathway and its associated molecules. Below are the molecules/targets related to other molecules in Akt pathway, click on them for product details.
Customized Services
Creative BioMart offers customized services to meet the specific needs of researchers. Whether it's custom protein expression, antibody development, or assay design, we can tailor our services to provide a personalized solution that meets the unique requirements of each project.
Technical Support
Creative BioMart emphasizes customer satisfaction and provides efficient customer support throughout the research process. Our dedicated customer service team is always available to answer inquiries, provide technical assistance, and address any concerns or questions researchers may have. We provide technical support in a comprehensive manner, such as online support, customer service hotline, technical documentation, etc., to help customers better use the company's products and services.
If you have any questions or requests, please feel free to contact us.
Why Choose Us?
Creative BioMart offers several unique advantages that make it an excellent choice for researchers studying the Akt pathway and its related molecules:
- Molecular Biology Expertise: With years of experience in the field of molecular biology, Creative BioMart has developed a wealth of expertise to provide researchers with cutting-edge solutions. Our team of scientists and technologists are well versed in the Akt pathway and can provide valuable insights and support to researchers in their studies.
- High Quality Products and Reliable Data: Creative BioMart is committed to providing the highest quality products. All products undergo a rigorous quality control process to ensure reliability and performance. We provide detailed product information, including specifications, applications, and validation data, so researchers can make informed decisions and have confidence in the reliability of the resources they receive.
- Continuous Innovation: Creative BioMart stays at the forefront of scientific innovation, regularly updating its products to keep up with the latest research trends and technological advances. By keeping abreast of the latest developments in the field, researchers have access to the most relevant and cutting-edge research resources.
- Collaborative Approach: Creative BioMart encourages collaboration and partnership with researchers around the world. By fostering collaborations, we aim to facilitate the exchange of knowledge, accelerate scientific discovery, and contribute to a deeper understanding of the Akt pathway and its impact on human health and disease.
Reference:
- Brito C, Barral DC, Pojo M. Subversion of Ras Small GTPases in Cutaneous Melanoma Aggressiveness. Front Cell Dev Biol. 2020;8:575223.