



Cell Biology
Creative BioMart Cell Biology Product List
Immunology Background
Background
Cell biology is a branch of biology and medicine. Cell biology uses microscopy and molecular biology methods to study cells to understand and elucidate biological processes at the cellular level. This includes the study of different cell compartments and cell organelles, cell division, movement of cells and cell groups, and communication between cells.
History
Cell biology, a cornerstone of modern life sciences, has its roots in the advent of microscopy. The discovery of cells is attributed to Robert Hooke in 1665, who observed the honeycomb-like structures in cork and coined the term "cell". This milestone was followed by the groundbreaking work of Antonie van Leeuwenhoek in the 1670s, who used his meticulously crafted microscopes to observe living cells, including bacteria and protozoa.
The 19th century marked a significant leap forward with the formulation of the cell theory by Matthias Schleiden and Theodor Schwann. This theory posited that all living organisms are composed of cells and that the cell is the fundamental unit of life. Rudolf Virchow later expanded this theory by emphasizing that all cells arise from pre-existing cells, a concept summarized in the phrase "Omnis cellula e cellula".
The 20th century saw an explosion of discoveries, driven by advancements in microscopy and biochemistry. The development of electron microscopy in the 1930s allowed scientists to explore cellular structures at unprecedented resolutions. Key discoveries, such as the double-helix structure of DNA by James Watson and Francis Crick in 1953, further illuminated the molecular basis of cellular function.
Cell types
There are two fundamental classifications of cells: prokaryotic and eukaryotic. Prokaryotic cells are distinguished from eukaryotic cells by the absence of a cell nucleus or other membrane-bound organelle.
Prokaryotic Cells
Prokaryotic cells, found in organisms such as bacteria and archaea, are simpler and generally smaller than eukaryotic cells. They lack membrane-bound organelles and a defined nucleus. The cell wall provides structural support and protection and is composed of peptidoglycan in bacteria, while archaea have different cell wall compositions, often lacking peptidoglycan. The plasma membrane is a phospholipid bilayer that regulates the movement of substances in and out of the cell. The cytoplasm is a gel-like substance that fills the cell and contains water, enzymes, nutrients, waste, and gases, and is the site of cellular processes such as metabolism and growth. The nucleoid is an irregularly shaped region that contains the cell's genetic material (DNA) and is not surrounded by a membrane. Ribosomes, which are the sites of protein synthesis, are smaller (70S) than eukaryotic ribosomes (80S). Plasmids are small, circular DNA molecules separate from the chromosomal DNA, often carrying genes beneficial for survival, such as antibiotic resistance. Flagella are long, whip-like structures used for motility and are composed of the protein flagellin. Pili (fimbriae) are hair-like structures on the surface of the cell, involved in adhesion to surfaces and conjugation (transfer of genetic material between cells). Some bacteria have an outer, gelatinous capsule layer, providing additional protection and aids in adhesion to surfaces.
Eukaryotic Cells
Eukaryotic cells are found in organisms such as plants, animals, fungi, and protists. They are more complex and typically larger than prokaryotic cells, containing membrane-bound organelles and a defined nucleus. The nucleus contains the cell's genetic material (DNA) organized into chromosomes and is surrounded by a nuclear envelope with nuclear pores for material exchange. The plasma membrane is a phospholipid bilayer with embedded proteins, carbohydrates, and cholesterol that regulates the movement of substances in and out of the cell. The cytoplasm consists of the cytosol and the organelles suspended in it, and it is the site of many metabolic activities. Mitochondria are the powerhouses of the cell, generating ATP through cellular respiration and having their own DNA and ribosomes. The endoplasmic reticulum (ER) exists in two forms: the rough ER, which is studded with ribosomes and involved in protein synthesis and modification, and the smooth ER, which lacks ribosomes and is involved in lipid synthesis and detoxification. The Golgi apparatus modifies, sorts, and packages proteins and lipids for transport. Lysosomes contain digestive enzymes to break down macromolecules, while peroxisomes are involved in lipid metabolism and detoxification of harmful substances. The cytoskeleton is a network of protein filaments (microfilaments, intermediate filaments, and microtubules) providing structural support and facilitating movement. Ribosomes in eukaryotic cells are the sites of protein synthesis and are larger (80S) than prokaryotic ribosomes (70S). Plant cells and some protists contain chloroplasts, which are the sites of photosynthesis and also have their own DNA and ribosomes. Vacuoles are large vesicles involved in the storage and maintenance of cell turgor pressure and are particularly prominent in plant cells. Centrosomes and centrioles are involved in microtubule organization and cell division. Plant cells, fungi, and some protists have a cell wall that provides structural support and protection, composed of cellulose in plants, chitin in fungi, and various other materials in protists.
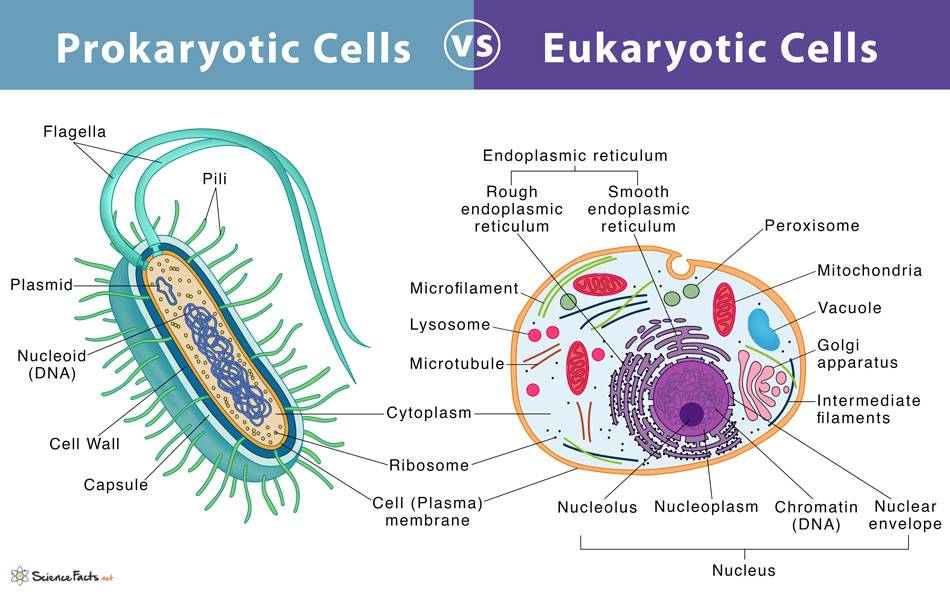 Fig. 1: Prokaryotic cells vs eukaryotic cells.
Fig. 1: Prokaryotic cells vs eukaryotic cells.Cell Metabolism
Cellular metabolism encompasses all the chemical reactions within a cell that are essential for sustaining life. These reactions are categorized as catabolic (breaking down molecules to release energy) and anabolic (synthesizing complex molecules from simpler ones).
Cellular respiration is a prime example of catabolic metabolism, involving glycolysis, the citric acid cycle, and oxidative phosphorylation. In glycolysis, glucose is converted to pyruvate, producing ATP and NADH. The citric acid cycle further oxidizes pyruvate, generating more NADH and FADH2. These electron carriers drive oxidative phosphorylation in the mitochondria, producing a significant amount of ATP.
Conversely, photosynthesis in plant cells is an anabolic process. It involves the conversion of light energy into chemical energy stored in glucose, through the light-dependent reactions and the Calvin cycle in chloroplasts.
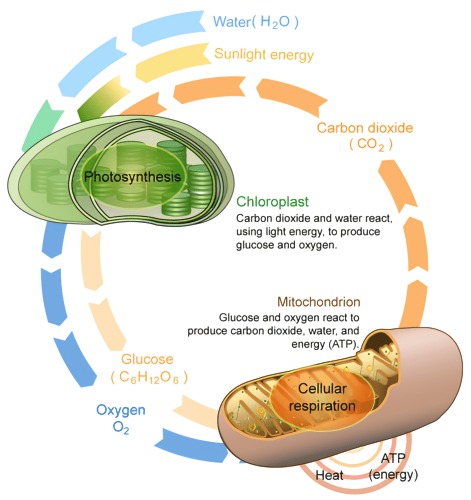 Fig. 2: Cellular respiration and photosynthesis.
Fig. 2: Cellular respiration and photosynthesis.Cell Division
Cell division is a fundamental process by which cells reproduce, ensuring the continuity of life and the maintenance of biological functions. This process is essential for growth, development, tissue repair, and reproduction in all living organisms. Cell division can occur through two main mechanisms: mitosis and meiosis, each of which serves a different purpose.
Mitosis
Mitosis is the process by which a single somatic cell divides to produce two genetically identical daughter cells. This process is essential for growth, development, and tissue repair in multicellular organisms. Mitosis consists of several stages: prophase, metaphase, anaphase, and telophase.
During prophase, chromatin condenses into visible chromosomes, each consisting of two sister chromatids joined at the centromere. The mitotic spindle, a microtubule structure, begins to form and the nuclear envelope breaks down. In metaphase, the chromosomes align at the equatorial plane of the cell, known as the metaphase plate, and spindle fibers attach to the centromeres. This is followed by anaphase, during which the sister chromatids are pulled apart by the spindle fibers toward opposite poles of the cell, ensuring that each new cell receives an identical set of chromosomes. In telophase, the chromosomes reach the poles, the nuclear envelope is re-formed around each set of chromosomes, and the chromosomes begin to decondense back into chromatin.
Mitosis is completed by cytokinesis, the division of the cytoplasm, resulting in two separate daughter cells. In animal cells, this process involves the formation of a cleavage furrow that splits the cell in two, whereas in plant cells, a cell plate forms along the equator of the cell that eventually develops into a dividing wall.
Meiosis
Meiosis, on the other hand, is a specialized form of cell division that produces gametes, or sex cells (sperm and egg), in sexually reproducing organisms. This process reduces the number of chromosomes by half, creating four genetically different haploid cells from one diploid cell, which is critical for maintaining the chromosome number of the species through generations. Meiosis involves two successive divisions: meiosis I and meiosis II.
In meiosis I, homologous chromosomes (each consisting of two sister chromatids) pair up and exchange genetic material through a process called crossing over during prophase I. This genetic recombination contributes to genetic diversity. The homologous pairs then align at the metaphase plate during metaphase I. In anaphase I, the homologous chromosomes are separated and pulled to opposite poles, in contrast to mitosis where sister chromatids are separated. In telophase I and subsequent cytokinesis, two haploid cells are formed, each containing one chromosome from each homologous pair.
Meiosis II resembles a mitotic division. During prophase II, the chromosomes condense again, and a new spindle apparatus forms. During metaphase II, the chromosomes align at the metaphase plate. During anaphase II, sister chromatids are separated and pulled to opposite poles. Finally, in telophase II and cytokinesis, the cells divide, resulting in four genetically distinct haploid gametes.
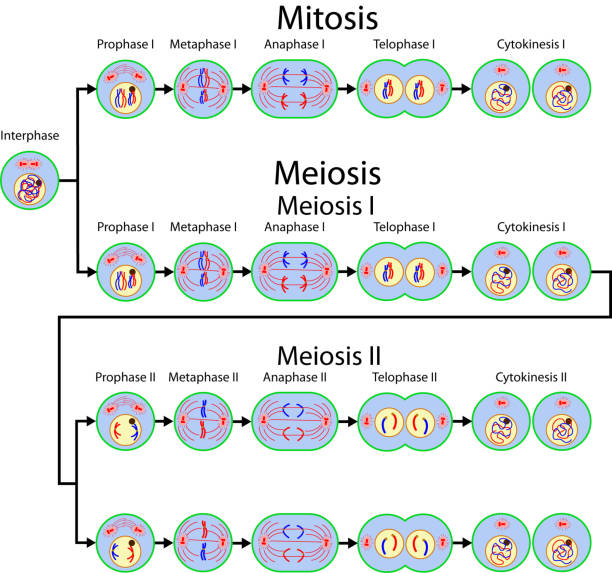 Fig. 3: Mitosis and meiosis cell division.
Fig. 3: Mitosis and meiosis cell division.Cell Migration
Cell migration is a fundamental biological process that involves the movement of cells from one location to another within an organism. This process is essential for various physiological functions, including embryonic development, wound healing, immune responses, and tissue regeneration. Cell migration is a highly coordinated and dynamic process, governed by intricate signaling pathways and interactions with the cellular environment.
Mechanisms of Cell Migration
Cell migration involves several key steps: protrusion, adhesion, translocation, and retraction.
Protrusion or extension begins when the cell extends its membrane forward to form structures such as lamellipodia (broad, sheet-like extensions) or filopodia (thin, spike-like protrusions). These structures are driven by the polymerization of actin filaments at the leading edge of the cell.
Adhesion occurs when the protrusions contact the extracellular matrix (ECM) or other cells through cell adhesion molecules such as integrins. These interactions provide traction for the cell to move forward.
Translocation is the forward movement of the cell body. The actin and myosin cytoskeleton generate contractile forces that pull the cell body in the direction of migration.
Retraction is the final step in which the back of the cell detaches from the substratum, allowing the cell to move forward. This process often involves the disassembly of focal adhesions and the retraction of the trailing edge of the cell.
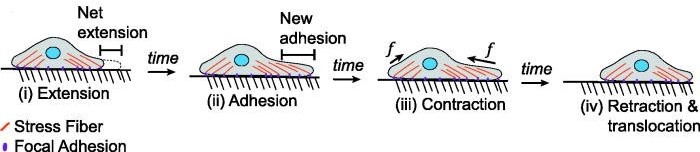 Fig. 4: Processes of mesenchymal migration typically involve a different process in which cells extend their leading edge and adhere, contract, and release their trailing edge.
Fig. 4: Processes of mesenchymal migration typically involve a different process in which cells extend their leading edge and adhere, contract, and release their trailing edge.Regulation of Cell Migration
Cell migration is tightly regulated by a complex network of signaling pathways and external cues. Key regulators include growth factors, chemokines, and ECM components.
Growth factors such as epidermal growth factor (EGF) and platelet-derived growth factor (PDGF) activate receptor tyrosine kinases (RTKs), which initiate downstream signaling cascades involving the MAPK/ERK and PI3K/Akt pathways. These pathways regulate cytoskeletal dynamics, adhesion turnover, and gene expression.
Chemokines are signaling molecules that create a gradient, guiding the directional movement of cells. Cells detect these gradients through chemokine receptors, which activate signaling pathways that direct cell migration towards higher concentrations of the chemokine.
Extracellular matrix (ECM) components provide structural support and biochemical signals that influence cell migration. Integrins, a family of cell surface receptors, mediate interactions between cells and the ECM, activating signaling pathways that regulate cytoskeletal reorganization and adhesion dynamics.
Physiological Roles of Cell Migration
Cell migration is critical for various physiological processes. During embryonic development, cell migration is essential for the formation of tissues and organs. Cells move to their proper locations, contributing to the intricate architecture of the developing organism. Wound healing relies on the migration of epithelial cells to cover and close the wound, as well as the migration of fibroblasts and immune cells to remodel tissue and fight infection.
In the immune response, cell migration enables immune cells to travel to sites of infection or injury, where they can fight pathogens and coordinate the immune response. Tissue regeneration also depends on cell migration as cells migrate to sites of injury to replace lost or damaged cells and restore tissue function.
Pathological Implications
While cell migration is essential for normal physiological processes, its dysregulation can contribute to various diseases. Cancer metastasis involves the migration of cancer cells from the primary tumor to distant sites, resulting in the spread of cancer throughout the body. Understanding the mechanisms of cell migration may aid in the development of therapies to prevent metastasis.
Chronic inflammation and autoimmune diseases can also result from aberrant cell migration. Immune cells may migrate inappropriately, leading to persistent inflammation and tissue damage. In addition, impaired cell migration can contribute to conditions such as impaired wound healing and fibrosis, where excessive migration and ECM deposition lead to tissue scarring.
Cell Signaling
Cell signaling is the process by which cells communicate with each other and respond to external stimuli. This communication is crucial for coordinating a wide array of cellular activities, including growth, differentiation, metabolism, and immune responses. Cell signaling involves the transmission of signals from the cell surface to the interior, ultimately resulting in a specific cellular response. The process can be divided into several key stages: signal reception, signal transduction, and cellular response.
Signal Reception
Signal reception begins when a signaling molecule, or ligand, binds to a specific receptor on the surface of the target cell. Receptors are usually proteins embedded in the cell membrane, though they can also be located inside the cell. There are various types of receptors, including:
- G-Protein-Coupled Receptors (GPCRs): These receptors interact with G-proteins to transmit signals from a variety of ligands, including hormones, neurotransmitters, and growth factors.
- Receptor Tyrosine Kinases (RTKs): These receptors have intrinsic enzymatic activity that phosphorylates specific tyrosine residues on the receptor itself and other target proteins, initiating a signaling cascade.
- Ion Channel Receptors: These receptors form channels that open or close in response to ligand binding, allowing specific ions to flow across the membrane, altering the cell's electrical state of the cell and triggering a response.
- Intracellular Receptors: Located within the cell, these receptors bind to ligands that can cross the plasma membrane, such as steroid hormones, leading to changes in gene expression.
Signal Transduction
Once a ligand binds to its receptor, the signal is transduced into the cell through a series of intracellular events, collectively known as a signaling cascade. This cascade often involves the activation of various proteins and second messengers that amplify and propagate the signal. Key components of signal transduction include:
- Second Messengers: Small molecules such as cyclic AMP (cAMP), calcium ions (Ca2+), and inositol trisphosphate (IP3) that transmit signals from receptors to target molecules within the cell.
- Protein Kinases: Enzymes that add phosphate groups to proteins (phosphorylation), which can activate or inhibit their function. Common types include serine/threonine kinases and tyrosine kinases.
- Adaptor Proteins: Proteins that facilitate the assembly of signaling complexes by linking various components of the signaling pathway.
Cellular Response
The final stage of cell signaling involves the cellular response, which can vary widely depending on the signal and the cell type. Responses may include:
- Changes in Gene Expression: Activation or repression of specific genes, leading to changes in protein synthesis and ultimately altering cellular function.
- Alterations in Metabolism: Activation or inhibition of metabolic pathways, affecting the cell's energy production and resource utilization.
- Changes in Cell Behavior: Modifications in cell shape, motility, or adhesion that are critical for processes such as immune responses, wound healing, and development.
- Programmed Cell Death (Apoptosis): Initiation of cell death in response to specific signals, which plays a critical role in development and disease prevention.
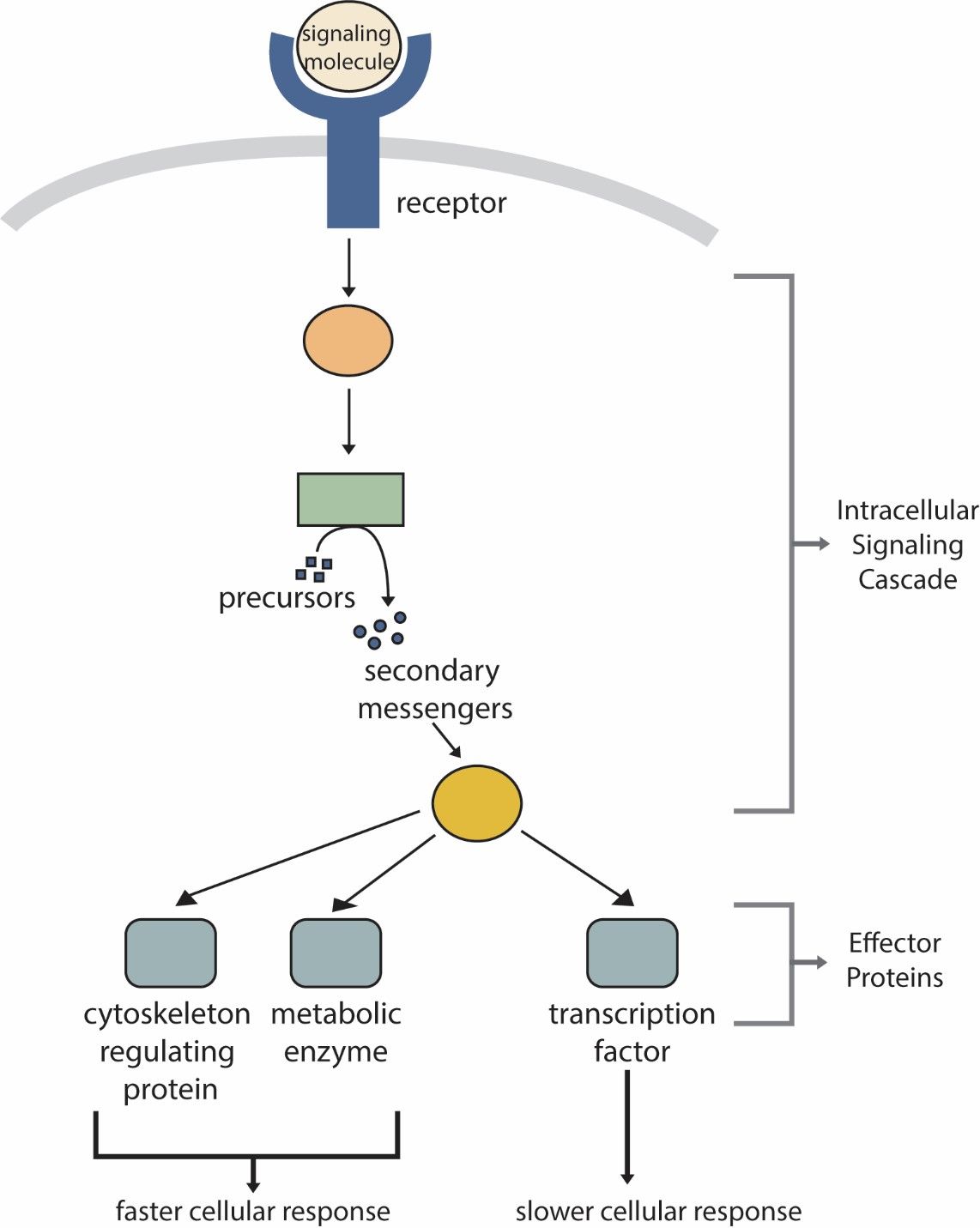 Fig. 5: Generic cell signaling pathway.
Fig. 5: Generic cell signaling pathway.Most important cell signaling pathways
- MAPK/ERK Pathway: The Mitogen-Activated Protein Kinase (MAPK) signaling pathway, also known as the Extracellular Signal-Regulated Kinase (ERK) signaling pathway, plays a crucial role in regulating cell growth, differentiation, and survival. It is activated by various growth factors, cytokines, and mitogens.
- PI3K/Akt Pathway: The Phosphoinositide 3-Kinase (PI3K)/Akt signaling pathway is crucial for regulating cell survival, growth, proliferation, and metabolism. It is activated by growth factors and insulin.
- JAK/STAT Pathway: The Janus Kinase (JAK)/Signal Transducer and Activator of Transcription (STAT) signaling pathway is important for transmitting information from extracellular cytokines to the nucleus, resulting in changes in gene expression. It is primarily involved in immune responses, hematopoiesis and inflammation.
- Wnt/β-Catenin Pathway: The Wnt/β-Catenin pathway is involved in the regulation of cell fate determination, proliferation, migration and embryonic development. It is activated by Wnt proteins that bind to Frizzled receptors.
Apoptosis
Apoptosis is a highly regulated and programmed process of cell death that is crucial for maintaining the health and stability of an organism. Unlike necrosis, which is a form of traumatic cell death resulting from acute cellular injury, apoptosis is a controlled and energy-dependent process that allows cells to die in a clean and orderly manner. This form of cell death is essential for various physiological processes, including development, tissue homeostasis, and the elimination of damaged or potentially harmful cells.
Mechanisms of Apoptosis
Apoptosis is initiated through two main pathways: the intrinsic (mitochondrial) pathway and the extrinsic (death receptor) pathway.
Intrinsic Pathway
The intrinsic pathway is regulated by the mitochondria and is typically activated by internal signals, such as DNA damage, oxidative stress, or other forms of cellular stress.
- Mitochondrial Outer Membrane Permeabilization (MOMP): This is a critical step in the intrinsic pathway, where the integrity of the mitochondrial outer membrane is compromised, leading to the release of pro-apoptotic factors like cytochrome c into the cytosol.
- Formation of the Apoptosome: Cytochrome c binds to Apaf-1 (apoptotic protease activating factor-1) in the cytosol, promoting the assembly of the apoptosome complex. These complex recruits and activates procaspase-9.
- Caspase Activation: Activated caspase-9 then cleaves and activates downstream effector caspases, such as caspase-3 and caspase-7, which carry out the cell death process by cleaving various cellular substrates.
Extrinsic Pathway
The extrinsic pathway is triggered by external signals, typically involving the binding of death ligands to their corresponding death receptors on the cell surface.
- Death Ligand Binding: Death ligands (such as Fas ligand or TNF-α) bind to their respective death receptors (such as Fas/CD95 or TNF receptor) on the cell membrane.
- Formation of the Death-Inducing Signaling Complex (DISC): This binding leads to the recruitment of adaptor proteins (such as FADD) and procaspase-8 to form the DISC.
- Caspase Activation: Within the DISC, procaspase-8 is cleaved and activated. Active caspase-8 can directly activate downstream effector caspases like caspase-3, leading to apoptosis.
Execution Phase
Once initiated by either pathway, the execution phase of apoptosis involves the activation of effector caspases, such as caspase-3, -6, and -7. These caspases cleave a variety of cellular proteins, leading to the characteristic morphological and biochemical changes of apoptosis:
- Cell Shrinkage and Rounding: Cells lose their adhesion to neighboring cells and the extracellular matrix, becoming more rounded and compact.
- Chromatin Condensation and DNA Fragmentation: Chromatin condenses, and DNA is cleaved into fragments by endonucleases, resulting in the characteristic "DNA ladder" pattern seen in apoptotic cells.
- Membrane Blebbing: The cell membrane forms protrusions known as blebs.
- Formation of Apoptotic Bodies: The cell fragments into smaller, membrane-bound vesicles called apoptotic bodies, which are then phagocytosed by neighboring cells or macrophages.
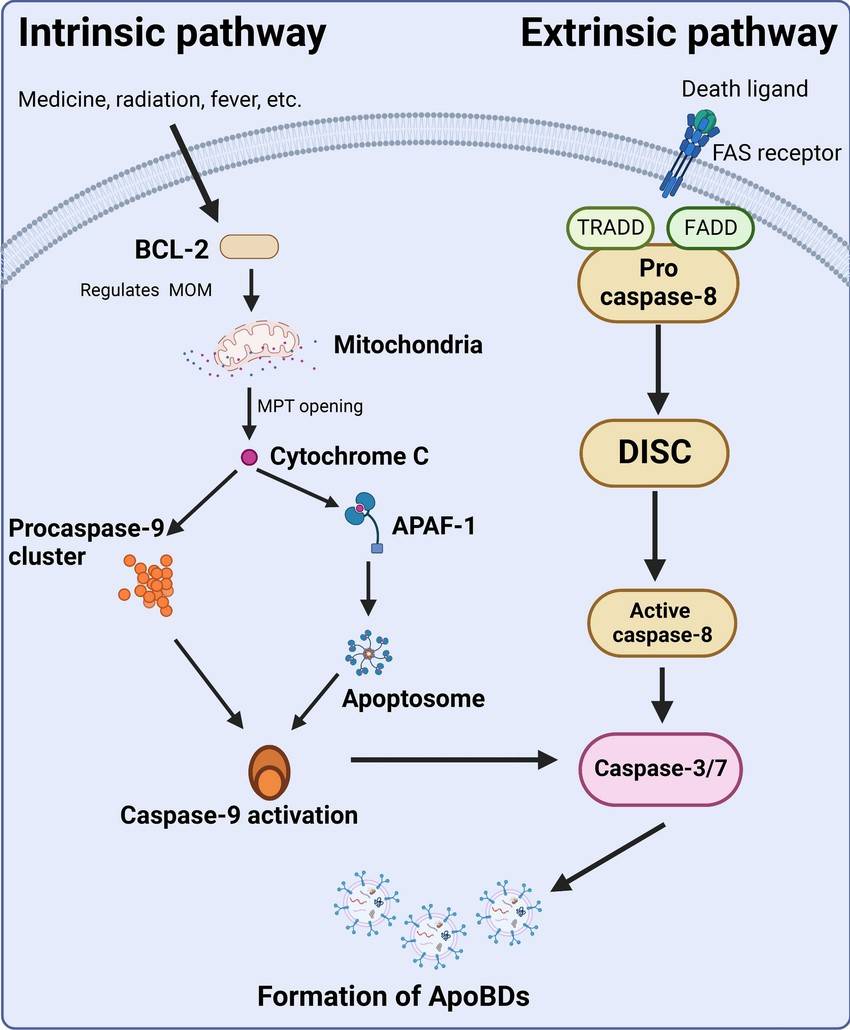 Fig. 6: Pathways involved in apoptosis.
Fig. 6: Pathways involved in apoptosis.Physiological Roles of Apoptosis
Apoptosis plays a crucial role in various physiological processes. During development, apoptosis shapes organs and tissues by eliminating excess or improperly positioned cells. In the immune system, apoptosis removes harmful or self-reactive immune cells, maintaining tolerance and preventing autoimmune diseases. Tissue homeostasis relies on apoptosis to balance cell proliferation and death, ensuring tissue integrity and function.
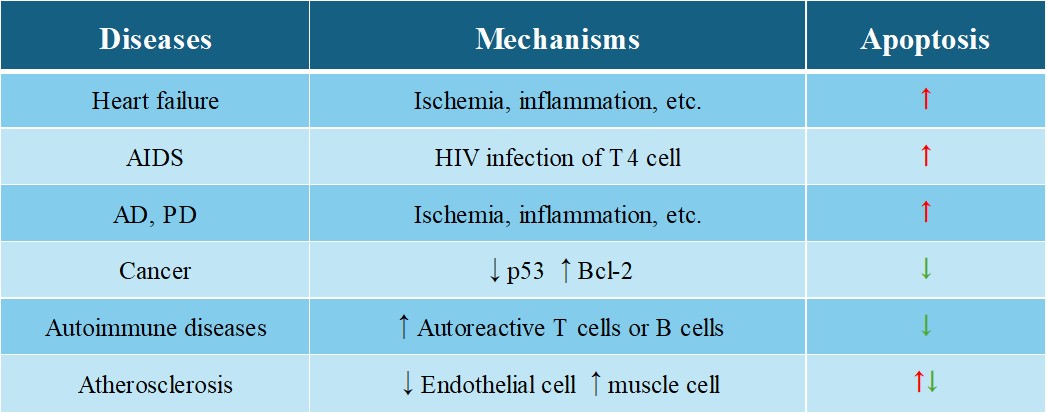
Pathological Implications
Dysregulation of apoptosis can lead to numerous diseases. Insufficient apoptosis can lead to uncontrolled cell proliferation and survival, contributing to the development of cancer. Many cancers have defects in apoptotic pathways that allow malignant cells to evade death and grow uncontrollably. Conversely, excessive apoptosis can cause degenerative diseases, such as neurodegenerative disorders (e.g., Alzheimer's, Parkinson's), in which excessive loss of neurons leads to functional decline. Autoimmune diseases can also result from defective apoptosis of immune cells, leading to the survival of self-reactive cells that attack the body's own tissues.
Tab. 1: Apoptosis-related diseases.
Case Study
Case 1: Salvador-Garcia, D.; et al. A force-sensitive mutation reveals a non-canonical role for dynein in anaphase progression. Journal of Cell Biology, vol. 223, no. 10, Oct. 2024.
As an important component of the microtubule network, the dynein motor plays a variety of roles in cell biology. In this study, researchers generated a series of missense mutations in the Drosophila dynein heavy chain. Their results show that dynein mutations associated with neurological disorders in humans lead to diverse problems, such as impaired cargo transport in nerve cells. In addition, they identify a novel mutation S3372C in the microtubule-binding domain that specifically blocks the metaphase-anaphase transition during mitosis in the embryo. This novel mutation specifically disrupts an embryonic function of dynein. The metaphase arrest and defects in centrosome assembly were confirmed by time-lapse analysis of S3372C mutant embryos in which components of the spindle apparatus were fluorescently labeled.
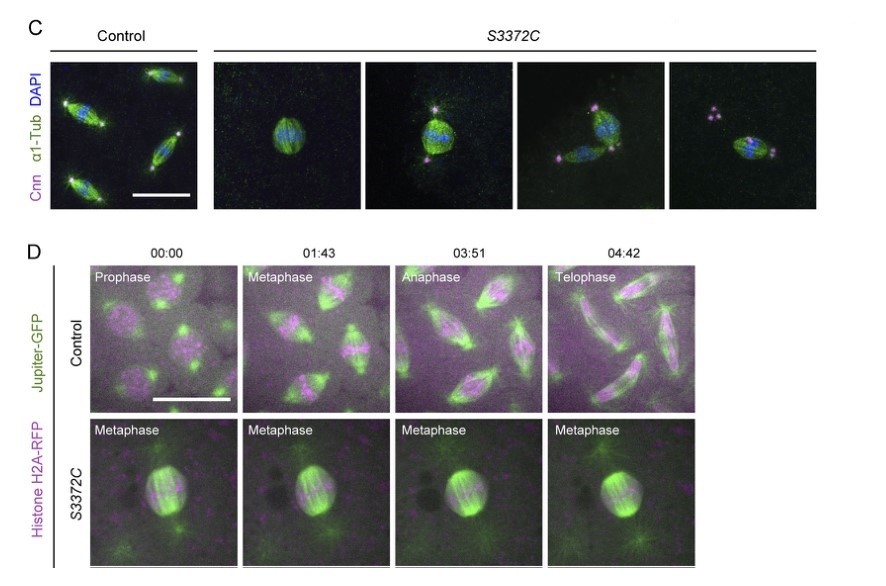 Fig. 7: S3372C causes metaphase arrest in the embryo. (C) Example confocal images of mitotic spindles in fixed control and S3372C embryos (Z-projection) stained with antibodies against centrosomin (Cnn; centrosome marker) and α1-tubulin (α1-Tub; microtubules) and DAPI. (D) Sample frames from time series (single focal plane) of control and S3372C embryos acquired during preblastoderm cycles. Jupiter-GFP and His2Av-mRFP label microtubules and chromatin, respectively. Time stamps are min:s. Scale bars: 20 µm.
Fig. 7: S3372C causes metaphase arrest in the embryo. (C) Example confocal images of mitotic spindles in fixed control and S3372C embryos (Z-projection) stained with antibodies against centrosomin (Cnn; centrosome marker) and α1-tubulin (α1-Tub; microtubules) and DAPI. (D) Sample frames from time series (single focal plane) of control and S3372C embryos acquired during preblastoderm cycles. Jupiter-GFP and His2Av-mRFP label microtubules and chromatin, respectively. Time stamps are min:s. Scale bars: 20 µm.Case 2: Xiang, JL.; et al. BAX-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proceedings of the National Academy of Sciences of the United States of America, vol. 93, no. 25, Dec. 1996, pp. 14559–63.
Apoptosis regulator BAX, also known as bcl-2-like protein 4, a member of the Bcl-2 gene family. BCL2 family members form hetero- or homodimers and act as anti- or pro-apoptotic regulators that are involved in a wide variety of cellular activities. Expression of BAX, in the absence of any other death stimulus, was sufficient to induce a common apoptotic pathway. This included activation of interleukin-1β-converting enzyme (ICE)-like proteases with cleavage of the endogenous substrates poly(ADP-ribose) polymerase and D4-GDI (GDP dissociation inhibitor for the rho family). The inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk) successfully blocked ICE-like protease activity and prevented FAS-induced death, but not BAX-induced death. Blocking ICE-like protease activity prevented nuclear and cytosolic substrate cleavage and DNA degradation following BAX induction. However, the decrease in mitochondrial membrane potential, production of reactive oxygen species, cytoplasmic vacuolation, and plasma membrane permeability downstream of BAX still occurred. Thus, BAX-induced changes in mitochondrial function and subsequent cell death do not appear to require the known ICE-like proteases.
In FAS-induced apoptosis, the fall in ΔΨm and ROS production are relatively late events and are blocked by zVAD-fmk, indicating that they lie downstream of ICE-like protease activation. In contrast, the reduction in ΔΨm and production of ROS induced by BAX continued in the absence of ICE-like protease activity when zVAD-fmk was present.
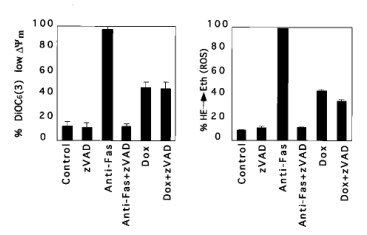 Fig. 8: Reduction of mitochondrial membrane potential ΔΨm and production of ROS after BAX induction. Jt-Bax cells were induced with Dox (1 μg/ml) or anti-FAS antibody (100 ng/ml) for 24 h in the presence or absence of 50 mM zVAD-fmk. The reduction of ΔΨm (Left) and ROS production (Right) were measured as described above.
Fig. 8: Reduction of mitochondrial membrane potential ΔΨm and production of ROS after BAX induction. Jt-Bax cells were induced with Dox (1 μg/ml) or anti-FAS antibody (100 ng/ml) for 24 h in the presence or absence of 50 mM zVAD-fmk. The reduction of ΔΨm (Left) and ROS production (Right) were measured as described above.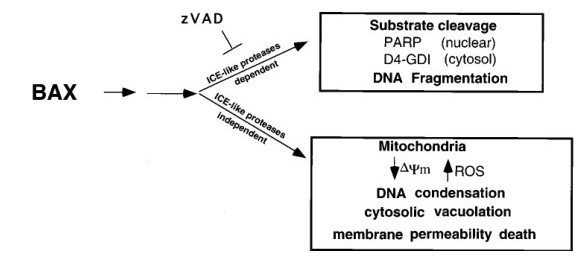 Fig. 9: Schematic model of BAX-induced apoptosis. The ICE-like protease-dependent and -independent pathways may be parallel or sequential.
Fig. 9: Schematic model of BAX-induced apoptosis. The ICE-like protease-dependent and -independent pathways may be parallel or sequential.References
- Alberts, Bruce. Molecular Biology of the Cell. Sixth edition, Garland Science, Taylor and Francis Group, 2015.
- Salvador-Garcia, David, et al. "A Force-Sensitive Mutation Reveals a Non-Canonical Role for Dynein in Anaphase Progression." Journal of Cell Biology, vol. 223, no. 10, Oct. 2024, p. e202310022. DOI.org (Crossref), https://doi.org/10.1083/jcb.202310022.
- Xiang, Jialing, et al. "BAX-Induced Cell Death May Not Require Interleukin 1β-Converting Enzyme-like Proteases." Proceedings of the National Academy of Sciences of the United States of America, vol. 93, no. 25, Dec. 1996, pp. 14559–63. PubMed Central, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC26172/.