
RNA Isolation protocol
RNA Isolation is frequently used in molecular biological experiments. The most important thing for isolate RNA is that you must protect RNA from degradation by RNAase. Here is a brief protocol to isolate RNA from leaf tissue.
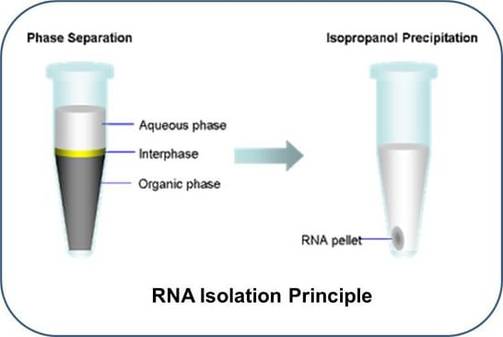
Fig. the schematic diagram of RNA isolation principle.
Materials
Leaf tissue
Liquid nitrogen
Extraction buffer
NaCl
β-mercaptoethanol
Chloroform
Isoamylalcohol
Isopropanol
Phenol
75% ethanol
Sterile water
Centrifuge
RNA Isolation protocol procedure
- 2 g of frozen tissue is ground to a fine powder with liquid nitrogen using mortar and pestle in presence of 2% insoluble polyvinylpyrollidone (PVPP).
- Powdered sample is transferred to a 50 ml sterile centrifuge tube, to which 20 ml pre-warmed extraction buffer at 65°C is added. Five molar NaCl (0.1 volume) and 1% β-mercaptoethanol to extraction buffer is added just before use.
- The sample tubes are vortexed for 1 min and incubated on ice for 5 min.
- Equal volume of chloroform: isoamylalcohol (24:1) is added, shake for 5 min and centrifuge at 14000 g for 15 min at 4°C.
- The supernatant is transferred to a fresh tube and added 0.1 X of 5 M NaCl, mixed gently, then added 1 X of cold isopropanol and finally precipitated overnight at - 20°C.
- The pellet is obtained by centrifuging at 14000 g for 20 min at 4°C.
- Pellet is washed with 75% ethanol air dried for 10 min and resuspended in 5 ml DEPC treated sterile water.
- Add equal volumes of Phenol:chloroform:isoamylalcohol (25:24:1) to the sample suspension and centrifuged at 14000 g for 15 min at 4°C.
- The supernatant is collected in a fresh centrifuge tube and add 0.1 X of 5 M NaCl, 1 X of cold isopropanol and precipitate overnight at -20°C.
- Pellet is obtained by centrifuging at 14000 g for 20 min at 4°C.
- The pellet is washed with 75% ethanol and resuspended in 500 ul DEPC treated sterile water.
- To the dissolved pellet, equal volumes of chloroform is added shook vigorously for 2 min and centrifuge at 12000 g for 15 min at 4°C and collect the supernatant.
- Add equal volumes of phenol:chloroform (1:1), mix vigorously for 5 min and centrifuge at 12000 g for 15 min at 4°C and collect the supernatant.
- Finally the supernatant is transferred to a fresh 1.5 ml sterile Eppendorf tube and add 0.1 V x 5 M NaCl, 1 X cold isopropanol. Precipitate overnight at -20°C.
- The pellet of RNA is obtained by centrifuging at 12000 g for 20 min at 4°C.
- After washing in 75% ethanol and air dried for 10 min, the pellet is resuspended in sterile water and the RNA sample obtained is stored at -80ºC.